Week 4 HW: Protein Design I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is roughly 20% protein by mass, so 500 g of meat contains about 100 g of protein. Since one Dalton equals 1 g/mol, an average amino acid at 100 Da means 100 g/mol. That gives 100 g ÷ 100 g/mol = 1 mole of amino acid residues, or about 6 × 10²³ amino acid molecules. So eating a steak hands you roughly Avogadro’s number of amino acids, a staggering count that just means “about a mole.”
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Digestion breaks dietary protein down to individual amino acids (or short peptides) in our gut, stripping away all the cow or fish specific information that lived in the sequence of those proteins. Once the amino acids are absorbed, they are just interchangeable building blocks, the same 20 letters every organism uses. Our ribosomes then assemble new proteins using our own mRNA templates, which come from our own DNA. So the food provides raw material, but your genome determines what gets built. Therefore, information is in the sequence, not the bricks. Thanks to that we are not turning to a fish!
Why are there only 20 natural amino acids?
The genetic code uses three nucleotide codons drawn from a 4 letter DNA alphabet, giving 4³ = 64 possible codons, comfortably enough to encode 20 amino acids with redundancy. Twenty also turns out to be enough chemical diversity to fold proteins and run all of biochemistry: you get charged residues, polar ones, hydrophobic ones, aromatics, and structurally special cases like proline, glycine, and cysteine. Beyond that, it is largely a frozen accident. Early life locked in this set, and the entire translation machinery (tRNAs, synthetases, ribosomes) co-evolved around it, so deviating became evolutionarily costly. A few exceptions exist (selenocysteine and pyrrolysine), showing the system is not completely rigid.
Can you make other non-natural amino acids? Design some new amino acids.
Yes, non-natural amino acids (ncAAs) are a major area of synthetic biology, pioneered by Peter Schultz and Jason Chin, who have expanded the genetic code to incorporate hundreds of new residues using engineered tRNA/synthetase pairs and reassigned stop codons. Existing examples include p-azido-phenylalanine (a click chemistry handle), photo-crosslinking benzophenone amino acids, fluorinated residues for ¹⁹F NMR, and phosphoserine for tunable phosphorylation. Some design ideas: a metal chelating residue (terpyridine side chain) for built-in catalysis; a photoswitchable azobenzene amino acid that lets light flip a protein between two conformations; a bioorthogonal alkyne residue for labeling; or a heavy tryptophan analogue with extra fused rings to deepen hydrophobic cores. The principle is that as long as the side chain does not break the backbone geometry the ribosome expects, you can design almost any chemistry into a protein.
Where did amino acids come from before enzymes that make them, and before life started?
Prebiotic chemistry. Amino acids form spontaneously under reasonable early Earth conditions. Stanley Miller and Harold Urey demonstrated this in 1953 by sparking a flask of methane, ammonia, hydrogen, and water; within days the flask contained glycine, alanine, and several other amino acids. Amino acids also rain down from space: the Murchison meteorite, which fell in Australia in 1969, carries over 70 amino acids including most of the 20 biological ones. Hydrothermal vents and mineral surfaces likely also drove abiotic synthesis. So before life, a primordial soup already had a pool of amino acids ready for early replicators (probably RNA based) to put to use.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left handed. Natural α-helices are right handed because they are built from L-amino acids, and the steric and torsional constraints of L chirality force the backbone into a right handed twist. D-amino acids are the exact mirror image, so a helix made entirely of them would be the mirror image of a natural one, left handed, with otherwise identical pitch, rise, and hydrogen bond geometry. This is well established and confirmed in synthetic D-protein chemistry.
Can you discover additional helices in proteins?
There are already several beyond the canonical α-helix: the 3₁₀-helix (tighter, 3 residues per turn, common at helix ends), the π-helix (wider, rarer), the left handed polyproline II helix (PPII, abundant in collagen and disordered regions), and the collagen triple helix itself. Coiled coil oligomers, dimers up through 7 helix bundles like in GPCRs (Thras showed this progression in lecture: laminin trimer, 4 helix bundles, the coronavirus spike’s 6 helix, 7 helix GPCRs), are higher order assemblies built from α-helices but with their own structural identities. With non-natural amino acids or β-amino acids you can build helices with completely different geometries (foldamers), so the catalogue is genuinely open ended.
Why are most molecular helices right-handed?
Because their building blocks are chiral and biology picked one chirality. L-amino acids have a specific spatial arrangement around their alpha carbon, and the allowed backbone dihedral angles (the φ/ψ region of the Ramachandran plot) heavily favor a right handed twist. A left handed α-helix made of L-amino acids is sterically strained and almost never seen. The deeper question of why life uses L-amino acids and not D ones is unresolved: it might be a frozen accident amplified from a tiny initial bias, possibly seeded by parity violation in the weak nuclear force or by chiral selection on mineral surfaces. Either way, once L was locked in, right handed helices followed automatically.
Why do β-sheets tend to aggregate?
A β-sheet has two edges that expose unsatisfied backbone hydrogen bond donors (N–H) and acceptors (C=O) sticking out into solvent. These edges are chemically sticky; the cheapest way to satisfy them is to recruit another β-strand to hydrogen bond against. Hydrophobic side chains on the sheet faces add a second driving force: they want to be buried, so two sheets stack face to face to exclude water. The result is that β-sheets greedily extend and stack, growing into multi-stranded assemblies, while α-helices can satisfy their hydrogen bonds internally and do not have this same edge problem.
What is the driving force for β-sheet aggregation?
Two cooperating forces. First, backbone hydrogen bonding: every additional strand zips a row of N–H···O=C bonds along the sheet edge, and these bonds are highly directional and energetically favorable. Second, the hydrophobic effect: water is entropically penalized when forced to organize around exposed hydrophobic side chains, so burying those side chains between stacked sheets releases water and increases entropy. Together these make β-sheet aggregates extraordinarily thermodynamically stable, which is exactly why amyloids resist heat, detergents, and proteases.
Why do many amyloid diseases form β-sheets?
Because the cross-β fold is a generic, sequence tolerant low energy sink that almost any polypeptide can fall into under the right conditions. The hydrogen bonds doing the heavy lifting are between backbone atoms, which every amino acid has, so the propensity is not unique to any one protein, it is a property of the polypeptide backbone itself. Once a small amyloid seed forms, it templates further misfolding (a prion like mechanism), so the process is self amplifying. That is why structurally unrelated proteins, Aβ in Alzheimer’s, α-synuclein in Parkinson’s, tau in tauopathies, prion protein in CJD, all converge on cross-β fibrils despite having totally different native folds.
Can you use amyloid β-sheets as materials?
Absolutely. Thras hit this in lecture when he compared spider silk (a β-sheet protein) to Kevlar, noting that humans engineered Kevlar inspired by the same backbone hydrogen bonding geometry that makes silk one of nature’s strongest materials. Functional amyloids exist naturally too: bacterial curli fibers form biofilm scaffolds, and mammalian Pmel17 organizes melanin in melanosomes. Engineered uses include self assembling peptide hydrogels for tissue scaffolding (Shuguang Zhang’s RADA16 is the canonical example), conductive amyloid nanowires, drug delivery fibrils, and adhesive coatings. The same property that makes amyloids pathological, extreme stability, makes them excellent biomaterials when you control the assembly.
Design a β-sheet motif that forms a well-ordered structure.
The trick is an amphipathic alternating pattern: hydrophobic and hydrophilic residues on alternating positions, so each β-strand has one hydrophobic face and one hydrophilic face. Shuguang Zhang’s RADA16, Ac-(RADA)₄-NH₂, the sequence Arg-Ala-Asp-Ala repeated four times, is the textbook example: alanines pack into a hydrophobic core, while alternating arginines (+) and aspartates (−) form complementary salt bridges on the polar face, locking the sheets into ordered nanofibers in water. A simple variant I would design: Ac-(VEVK)₄-NH₂, where valine gives the hydrophobic face and alternating glutamate/lysine give a charge zipper polar face; this should self assemble into stable antiparallel β-sheets at neutral pH. To enforce well ordered (rather than messy aggregated) structure, cap the ends with proline or glycine to prevent runaway elongation, and keep the strand length around 8 to 16 residues so a single sheet can register cleanly without frame shifts.
Part B. Protein Analysis and Visualization
For this assignment, I selected Neuroligin-4X (NLGN4X) from human (Homo sapiens). I chose this protein because it has both sequence information and an experimentally solved 3D structure, which makes it suitable for practicing a protein analysis workflow. NLGN4X is also related to synaptic biology, so it is a useful example for connecting protein structure to a biological context. The main sequence resource I used is UniProt Q8N0W4, and the main structure resource is RCSB PDB 3BE8, titled “Crystal structure of the synaptic protein neuroligin 4.” The RCSB entry identifies the protein as human Neuroligin-4, X-linked, and links it to the gene name NLGN4X and the UniProt accession Q8N0W4. NCBI also describes NLGN4X as a neuronal cell-surface protein. This background gives the protein some biological relevance, but the main goal of this report is to analyze its sequence and structure using online databases and PyMOL.
For the sequence analysis, I used the full-length human NLGN4X sequence from UniProt. The full protein is 816 amino acids long. For the structure analysis, I used PDB 3BE8. This structure does not contain the entire full-length protein; instead, it contains the extracellular cholinesterase-like domain of NLGN4X. This means that the full UniProt sequence is used for the amino acid sequence and frequency analysis, while the PDB structure is used for 3D visualization. The sequence of the crystallized construct can also be viewed through the RCSB FASTA page for 3BE8.
The full-length amino acid sequence of human NLGN4X is shown below in FASTA format:
After counting the amino acids in the full-length sequence, the most frequent amino acid is leucine (L), which appears 75 times. The next most common amino acids are threonine (T), which appears 72 times, and proline (P), which appears 61 times. The full amino acid count is: L = 75, T = 72, P = 61, G = 50, D = 50, V = 47, A = 46, I = 46, N = 45, S = 44, E = 37, K = 36, Y = 36, Q = 35, R = 32, F = 32, H = 27, M = 22, W = 13, and C = 10. This result shows which amino acids are most common in the sequence, but amino acid frequency alone should not be over-interpreted because it does not fully explain the protein’s structure or function.
To search for sequence homologs, the NLGN4X sequence can be submitted to the UniProt BLAST tool. A reasonable approach is to paste the full FASTA sequence into UniProt BLAST, search against UniProtKB, and record significant matches using a cutoff such as E-value < 1e-5. The results are expected to include NLGN4X orthologs from other species and related human neuroligin proteins such as NLGN1, NLGN2, NLGN3, and NLGN4Y. The exact number of homologs may vary depending on the BLAST settings and database version, so this number should be filled in after running the search:
NLGN4X belongs to the neuroligin protein family. Neuroligins are cell-surface proteins involved in synaptic adhesion. NLGN4X also contains a cholinesterase-like extracellular domain and is related to the type-B carboxylesterase/lipase protein family. Although the fold resembles some enzyme families, NLGN4X is generally discussed as a synaptic adhesion protein rather than as a classical enzyme.
The RCSB structure used for this report is PDB ID 3BE8. This structure was solved by X-ray diffraction and has a resolution of 2.20 Å. In protein crystallography, smaller resolution values usually indicate better structural detail. Since the assignment describes 2.70 Å as a good-resolution cutoff, the 2.20 Å resolution of 3BE8 is good for this project. The structure was deposited on 2007-11-16 and released on 2008-01-29.
The solved structure contains the Neuroligin-4X extracellular domain as a homodimer, meaning that two copies of the protein domain are present together. The structure also contains some non-protein molecules, including N-acetylglucosamine (NAG) sugar residues and small crystallographic molecules or ions such as citrate and phosphate. Therefore, the structure is mainly a protein dimer, but it also includes additional molecules that are not part of the protein chain.
In terms of structural classification, NLGN4X can be described as a cell-adhesion protein with an alpha/beta hydrolase-like or cholinesterase-like fold. This type of fold contains both alpha helices and beta sheets. For this report, I describe NLGN4X as a neuroligin family protein with a mixed alpha/beta structural fold.
I visualized the structure using PyMOL. First, I loaded the structure with the following command:
The cartoon view was generated using:
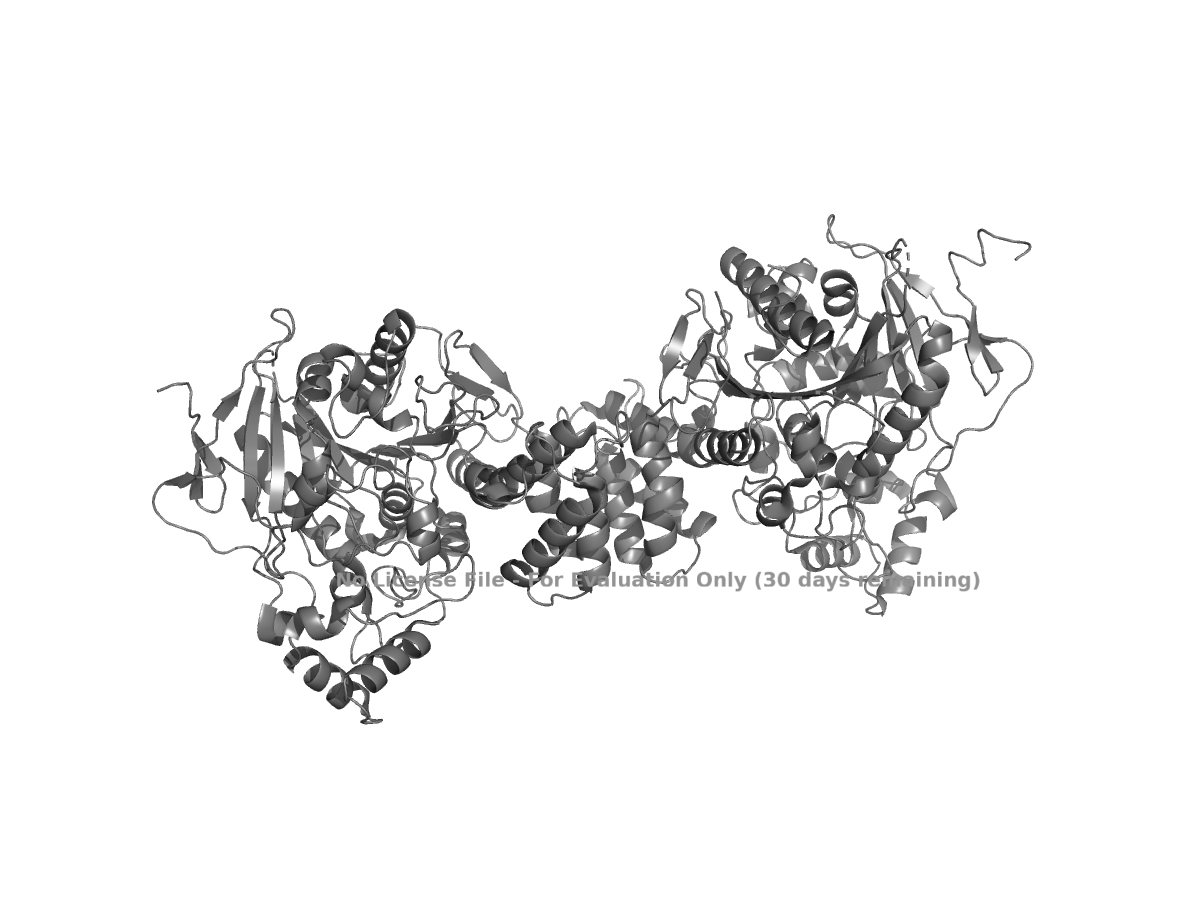
Figure 1. Cartoon view of human NLGN4X from PDB 3BE8.
The ribbon view was generated using:

Figure 2. Ribbon view of human NLGN4X from PDB 3BE8.
The ball-and-stick view was generated using:
Because showing the entire protein in ball-and-stick style can look crowded, a close-up region is usually easier to interpret.
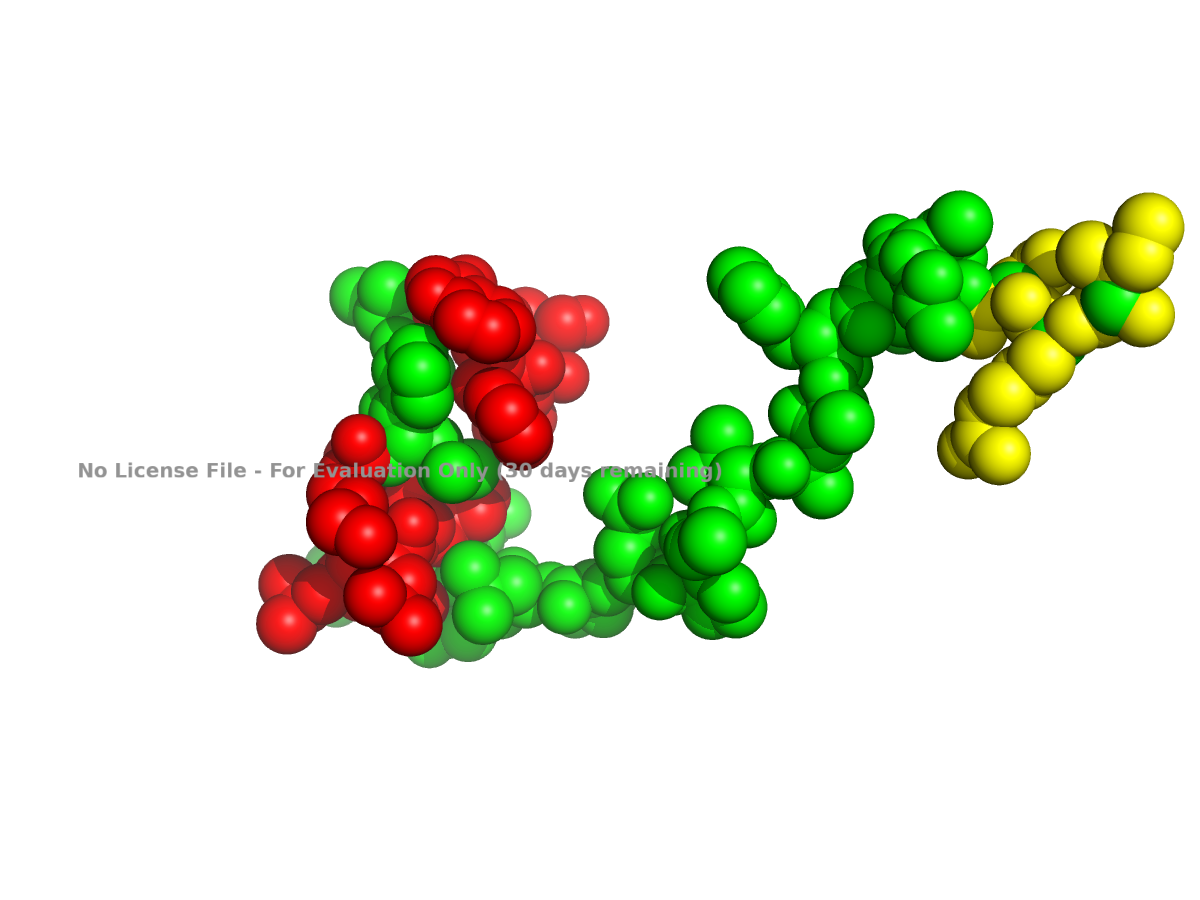
Figure 3. Ball-and-stick close-up view of part of human NLGN4X.
I also colored the protein by secondary structure in PyMOL. In this view, alpha helices are colored red, beta sheets are colored yellow, and loops are colored green:
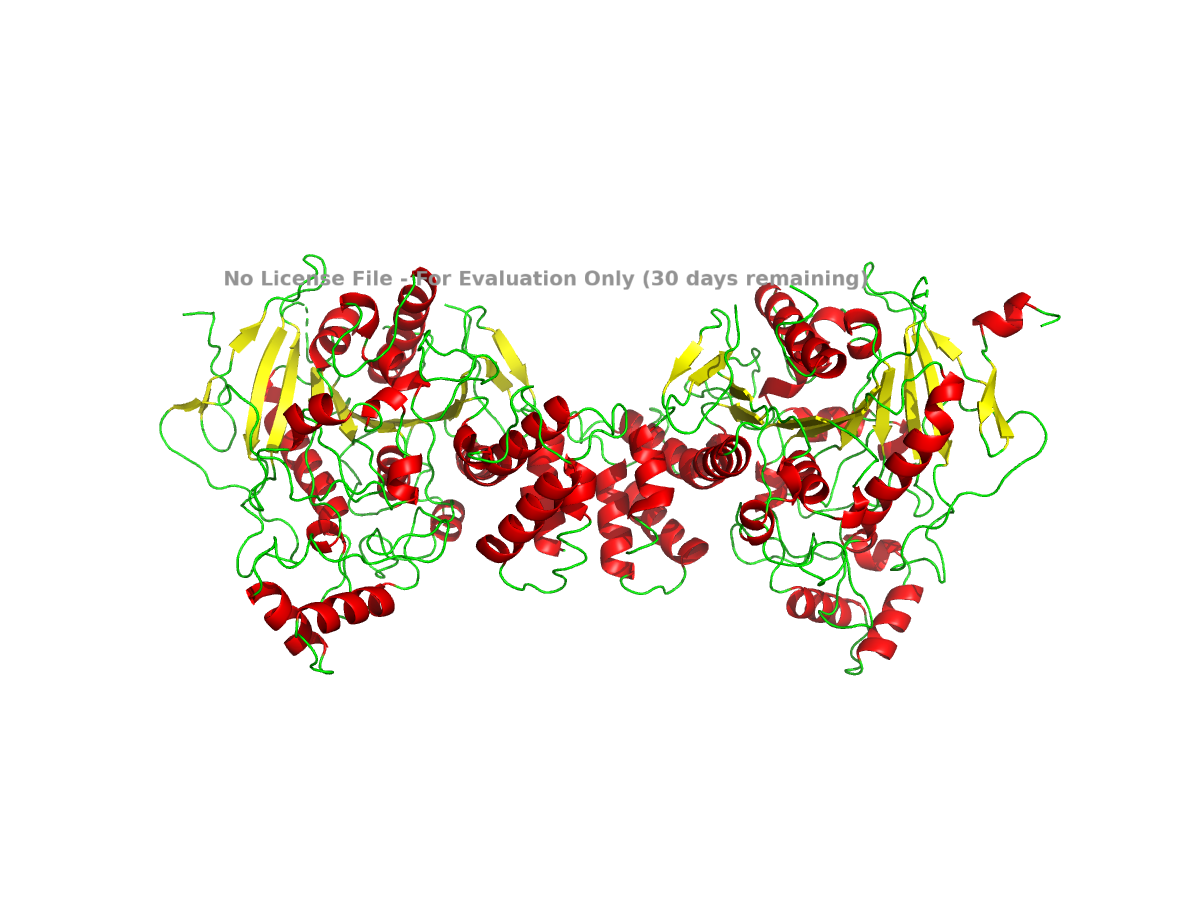
Figure 4. NLGN4X colored by secondary structure. Helices, sheets, and loops are shown in different colors.
Based on this view, NLGN4X appears to be a mixed alpha/beta protein. It is not purely helical or purely beta-sheet. The structure contains a beta-sheet-containing core along with several alpha-helical regions around it.
To examine residue type distribution, I colored hydrophobic residues orange and hydrophilic or polar/charged residues cyan:
In this view, hydrophobic residues are expected to appear more often in buried parts of the structure, while hydrophilic residues are more likely to appear on the surface where they can interact with water. This is a general pattern for many soluble or extracellular protein domains. For this protein, I would use the visualization to check whether this pattern is visible rather than treating it as a definitive conclusion from the sequence alone.
Finally, I used a surface representation to look for grooves, depressions, or possible pocket-like regions:
For a clearer surface, I used:
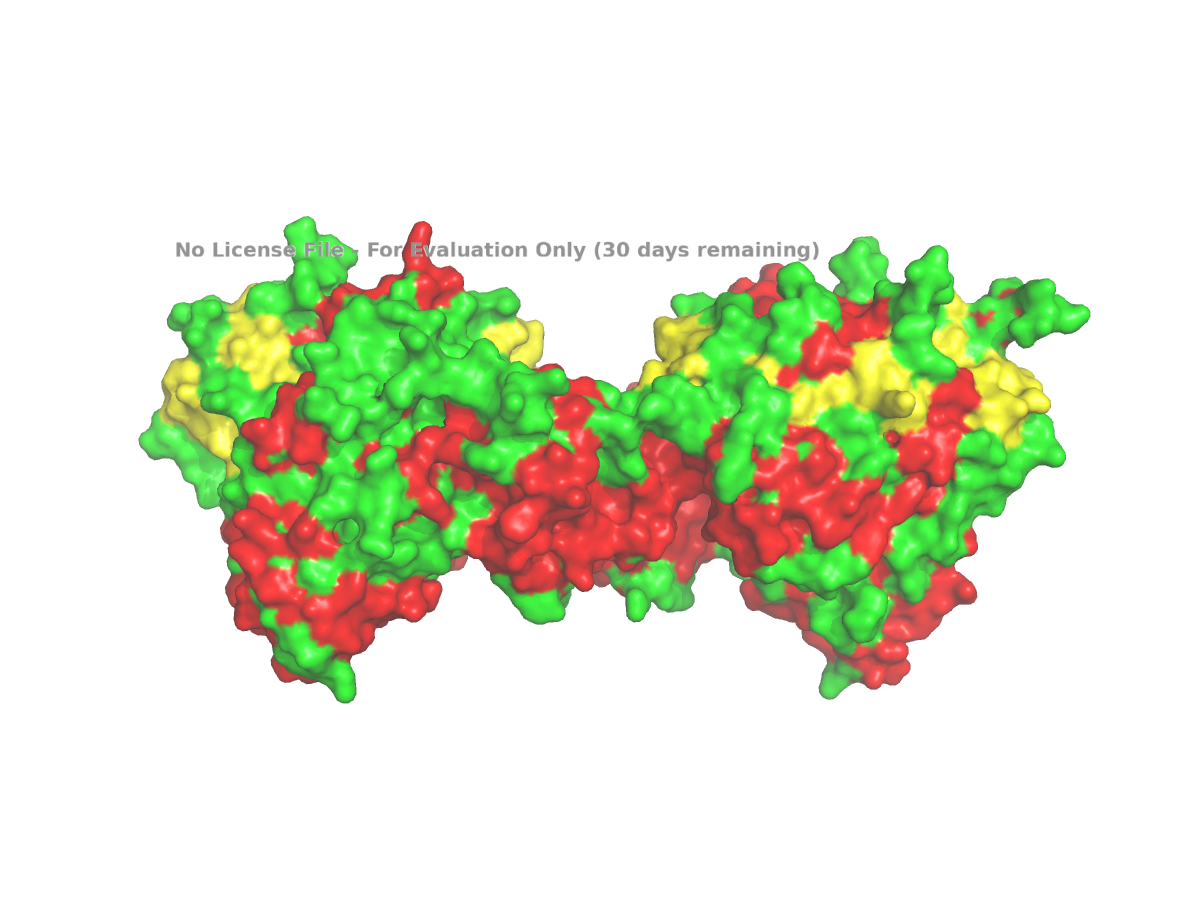
Figure 5. Surface view of human NLGN4X from PDB 3BE8, used to inspect grooves or possible pocket-like regions.
The surface view shows that the protein is not completely smooth. There appear to be grooves and depressions on the surface. These can be described as possible pocket-like surface features, although I would avoid calling them enzyme active sites unless there is specific evidence for that. In this assignment, the surface view is mainly used to make a visual observation about the shape of the protein.
Overall, human NLGN4X is a suitable example for this assignment because its sequence and structure are available in standard databases. The full-length protein is 816 amino acids long, and leucine is the most frequent amino acid in the sequence. The selected structure, PDB 3BE8, represents the extracellular domain of NLGN4X and was solved by X-ray diffraction at 2.20 Å, which is better than the 2.70 Å cutoff given in the prompt. The structure shows a mixed alpha/beta fold, and PyMOL can be used to examine the protein in cartoon, ribbon, ball-and-stick, secondary-structure, residue-type, and surface views.
## Part C. Using ML-Based Protein Design Tools
I chose Thaumatin I from Thaumatococcus daniellii because it is a food-related sweet-tasting protein with a well-characterized 3D structure. Its sequence is available in UniProt P02883, and its structure is available as PDB 1RQW, with the corresponding RCSB FASTA sequence. UniProt lists Thaumatin I as 235 amino acids long, and RCSB reports the 1RQW structure as an X-ray structure with 1.05 Å resolution.
C1. Protein Language Modeling
Deep Mutational Scans
The heatmap shows the ESM2 deep mutational scan for thaumatin. Each column represents a position in the protein sequence, and each row represents a possible amino acid substitution. Brighter colors correspond to higher LLR scores, meaning the model finds that mutation more compatible with the sequence context, while darker colors correspond to lower LLR scores. Overall, the heatmap shows that some regions are more tolerant to substitutions, while other regions contain many low-scoring mutations and may be more constrained.
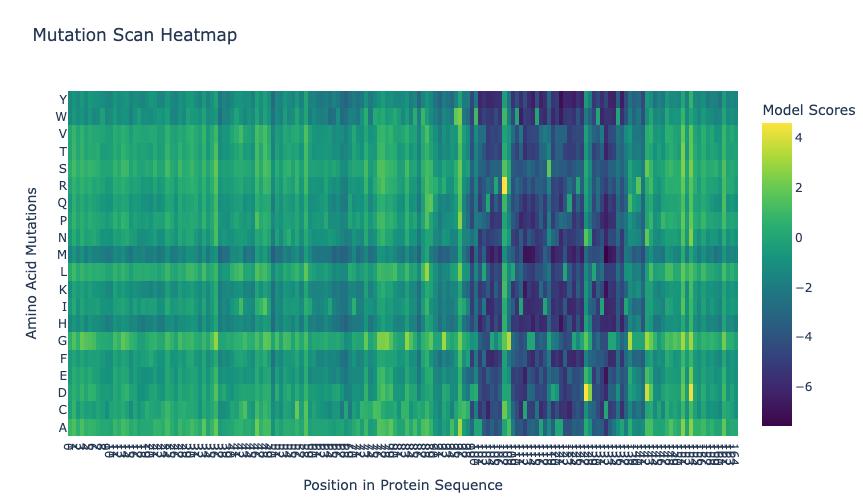
In the ESM2 mutational scan, the most positive mutation score was for T2M, where threonine at position 2 is replaced by methionine. This mutation had an LLR score of 4.8903, meaning that the model scored methionine at this position as more likely than the original threonine. Since threonine is polar and methionine is larger and hydrophobic, this result may suggest that ESM2 finds a more hydrophobic residue compatible at this position. However, this should be interpreted cautiously because the scan is based on language-model likelihoods, not an experimental measurement of thaumatin stability or sweetness.
#### Latent Space Analysis
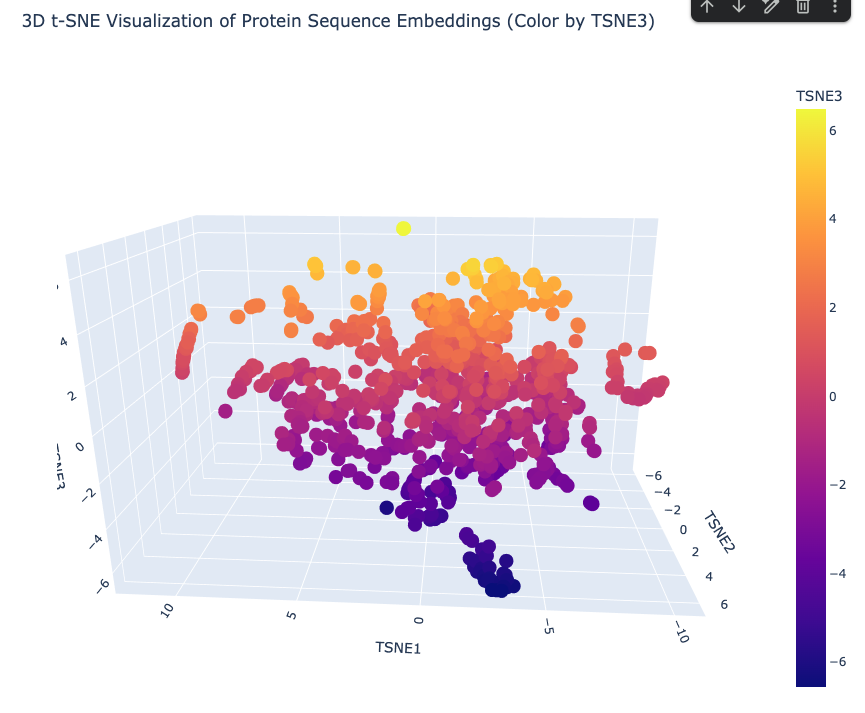
The latent-space analysis represents protein sequences as embeddings and then reduces them into three t-SNE dimensions for visualization. In the 3D t-SNE visualization of protein sequence embeddings, each point represents a protein sequence, and the axes represent reduced embedding dimensions. The plot shows several neighborhoods of proteins, suggesting that the embedding captures sequence-level similarities. Because the points are not distributed as one uniform cloud, the model appears to separate proteins into local groups based on features such as sequence similarity, domain composition, amino acid patterns, or protein family. I interpreted this plot as a qualitative map of protein similarity rather than an exact measurement of structural or functional relationships.