Week 6 HW: Genetic Circuits I
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion HF PCR Master Mix is a ready-to-use 2X solution that contains everything needed for PCR except the template, primers, and water.
The most important component is Phusion DNA Polymerase, which is the enzyme that copies DNA during the reaction. The standard alternative is Taq polymerase, a widely used enzyme isolated from a heat-resistant bacterium called Thermus aquaticus. Taq survives the high temperatures of PCR but has no proofreading ability, so it cannot correct mistakes it makes while copying, resulting in a relatively high error rate. Phusion addresses this by including a proofreading domain that catches and fixes errors as they occur, making it about 50 times more accurate than Taq. This accuracy is important in a mutagenesis experiment where only the specific, intended mutations should be introduced and any unintended errors would compromise the result.
The mix also contains dNTPs, which are the four DNA building blocks (dATP, dTTP, dGTP, dCTP) that the polymerase uses to construct new strands. MgCl₂ is included as well, providing magnesium ions that the polymerase requires to function and that also affect how tightly primers bind to the template. Finally, the HF Buffer maintains the right pH and salt levels so the enzyme works reliably across different templates.
2. What are some factors that determine primer annealing temperature during PCR?
The annealing temperature is typically set 2 to 5°C below the melting temperature (Tm) of the primer pair. The Tm is the temperature at which half of the primer-template bonds break apart, so setting the annealing temperature just below it ensures primers bind firmly without sticking to the wrong locations.
Several factors affect Tm. Primer length matters because longer primers form more bonds with the template and therefore require more heat to detach, resulting in a higher Tm. GC content also plays a role since G-C base pairs form three hydrogen bonds while A-T pairs form only two, meaning primers with more G and C bases bind more tightly and have a higher Tm.
Salt concentration in the buffer raises the effective Tm because magnesium and other ions stabilize the DNA duplex. Mismatches lower the Tm because they weaken binding. When a primer contains intentional mismatches, it grips the template less tightly, so a lower annealing temperature is needed to allow binding to occur. Primer concentration and secondary structures such as hairpins can also shift the effective annealing behavior.
3. Compare and contrast PCR and restriction enzyme digests as methods for producing linear DNA fragments.
Both PCR and restriction enzyme digestion produce linear DNA fragments, but they work through different mechanisms and are suited to different situations.
In terms of protocol, restriction digestion is the simpler method. The DNA is mixed with the enzyme in an appropriate buffer and incubated at 37°C for about an hour. PCR is more involved and requires primer design, a reaction setup with polymerase and dNTPs, and a thermocycling program that moves through denaturation, annealing, and extension steps, taking around 90 minutes in total.
The two methods also differ in how flexible they are. Restriction enzymes can only cut at their specific recognition sequences, so the location of the cut is fixed by whatever sites exist in the template. PCR can amplify any region defined by the primer binding sites, giving full control over fragment boundaries. PCR also amplifies the DNA exponentially from a very small starting amount, while restriction digestion simply cuts the DNA that is already present without increasing the quantity.
A key advantage of PCR is the ability to introduce mutations through mismatches built into the primers. The amplified product will carry those changes. Restriction enzymes cannot introduce new sequence and can only cut existing DNA.
| PCR | Restriction Enzyme Digest |
|---|---|
| Requires primer design and a thermocycling program. More involved setup. | Simpler protocol. Mix with enzyme and buffer, incubate at 37°C. |
| Fragment boundaries are fully flexible, defined by primer placement. | Can only cut at specific recognition sequences already in the DNA. |
| Primers can introduce mutations into the product. | Cannot change sequence, only cuts existing DNA. |
| Amplifies DNA exponentially from very small amounts. | Does not amplify, only cuts what is already present. |
| Best when custom boundaries, mutations, or overhangs are needed. | Best when convenient cut sites already exist and no changes are needed. |
In this lab PCR is the right choice because the chromophore mutations need to be introduced and overlapping ends need to be added for Gibson assembly, neither of which restriction digestion can do.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The most important requirement is that the fragments have overlapping ends of around 20 to 40 base pairs. These overlaps are what allow the fragments to recognize and stick to each other during Gibson assembly. When designing PCR primers, the overlapping regions need to be built into the primer sequences so that the amplified fragment carries them at its ends. The overlaps must also match the correct neighboring fragment so everything assembles in the right order.
Unlike restriction cloning, Gibson assembly does not require specific cut sites in the DNA. However, the ends must be carefully designed to be complementary to adjacent fragments. After PCR or digestion, the DNA should also be clean and free of contaminants so the assembly enzymes can work efficiently. Running a gel to confirm the fragments are the expected size is a useful check before proceeding to the assembly step.
5. How does plasmid DNA enter E. coli cells during transformation?
Plasmid DNA enters E. coli by making the cell membrane temporarily permeable so the DNA can pass through. There are two common methods for doing this.
The first is heat-shock transformation. The cells are treated with calcium chloride, which neutralizes the negative charges on both the DNA and the membrane. This reduces the natural repulsion between them and allows the DNA to sit against the cell surface. A sudden heat shock at 42°C then causes the membrane to expand rapidly, opening temporary pores. The DNA diffuses through those pores into the cell. The cells are immediately cooled so the membrane reseals, and then given nutrients and time to recover before being plated on selective media.
The second method is electroporation. Instead of heat, a short electrical pulse is applied to the cells. This creates tiny pores in the membrane through which the plasmid DNA can pass. Electroporation is generally more efficient than heat shock and is commonly used when transformation efficiency needs to be very high.
6. Describe another assembly method in detail: Golden Gate Assembly
Golden Gate Assembly is a one-pot cloning method that uses Type IIS restriction enzymes, most commonly BsaI, to join multiple DNA fragments in a single reaction without leaving any scar sequence at the junctions.
The key property of Type IIS enzymes is that they cut outside their recognition sequence rather than within it. This means the recognition site can be placed at the end of a fragment so that when the enzyme cuts, the recognition site itself is removed. What remains is a short custom 4-base-pair overhang at each junction, which the designer specifies in the primer sequence. Because every junction in the assembly has a different 4-base overhang, each fragment can only join its correct neighbor in the correct orientation. Order and direction are determined by the overhang sequences, not by chance.
The reaction contains all fragments, BsaI, and T4 ligase together in one tube. It cycles between 37°C, where the enzyme cuts, and 16°C, where the ligase seals compatible overhangs. Once two fragments are correctly joined, the BsaI recognition site is gone from that junction and the ligated product cannot be re-cut. This drives the reaction toward the fully assembled product over multiple cycles.
Compared to Gibson assembly, Golden Gate offers more precise control over junction sequences and scales well to 10 or more fragments in a single reaction. Gibson is simpler for small assemblies of 2 to 4 fragments because it only requires sequence overlap in the primers and runs isothermally at 50°C. Golden Gate requires that the BsaI recognition sequence does not appear anywhere inside the fragments being assembled, otherwise the enzyme will cut in the wrong place.
| Golden Gate | Gibson | |
|---|---|---|
| Overhang type | 4-bp sticky ends, custom designed | ~25 bp single-stranded overlap |
| Reaction temperature | Cycles between 37°C and 16°C | Isothermal at 50°C |
| Scarless junctions | Yes | Yes |
| Best for | 10 or more fragments, ordered assembly | 2 to 4 fragments, simple workflow |
| Requires RE site in primers | Yes | No |
For this chromophore lab, Gibson is the more practical choice because only two fragments need to be joined. Golden Gate becomes the better option when assembling many parts in a defined order, such as in metabolic pathway engineering or combinatorial library construction.
Benchling
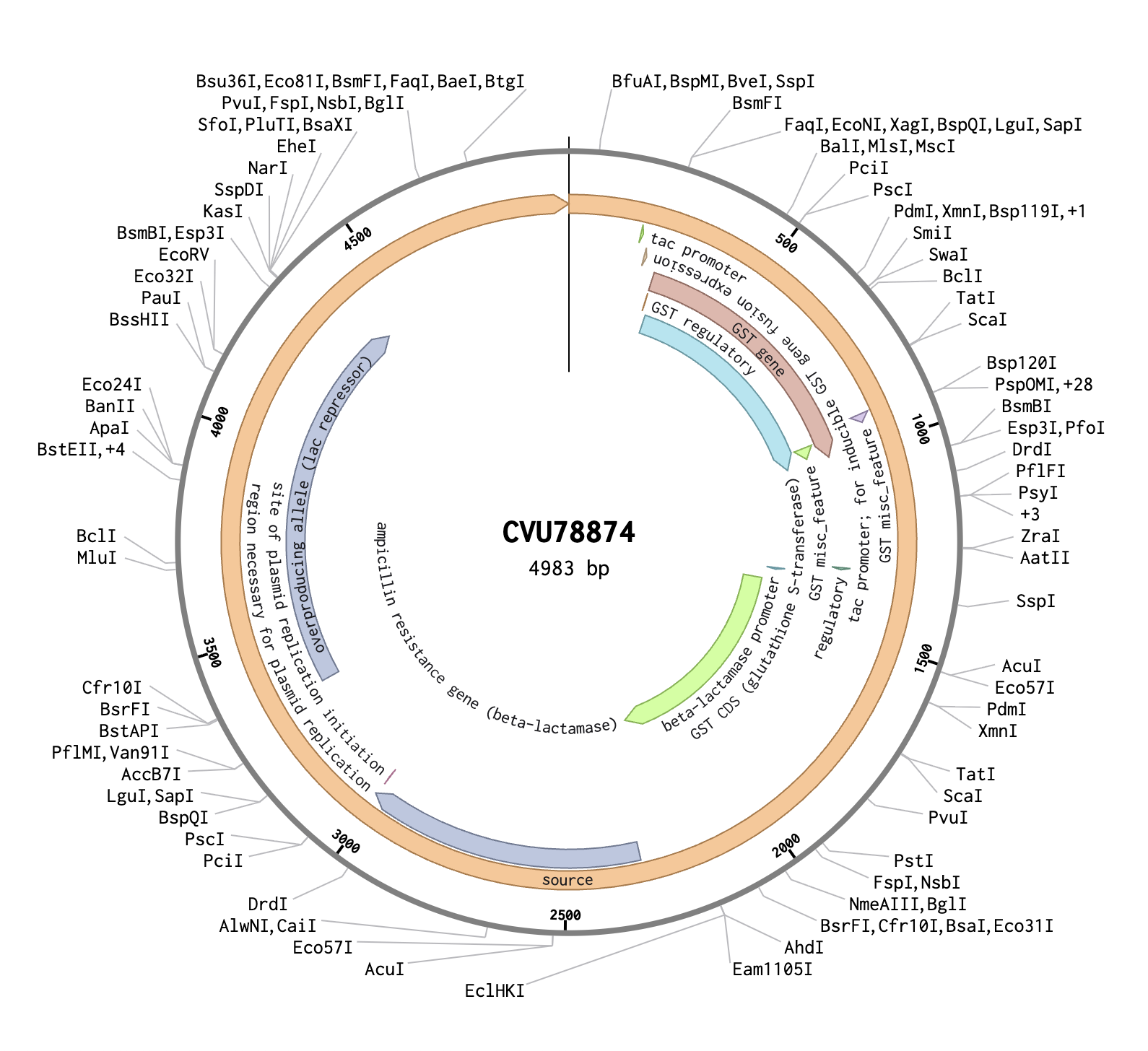
In Benchling, I created a new DNA sequence (type: DNA, topology: circular) to represent the pGEX plasmid. I imported it from the NCBI database using the accession number CVU78874, and confirmed it to be 4983 bp. The plasmid map showed several key annotated regions: the GST CDS (glutathione S-transferase), which is the orange region on the map and acts as a fusion tag so the inserted protein can be purified later using glutathione beads; the tac promoter, which drives expression of the insert and is switched on by adding IPTG to the growth media; and the ampicillin resistance gene, which allows only bacteria that successfully took up the plasmid to survive on selection plates.
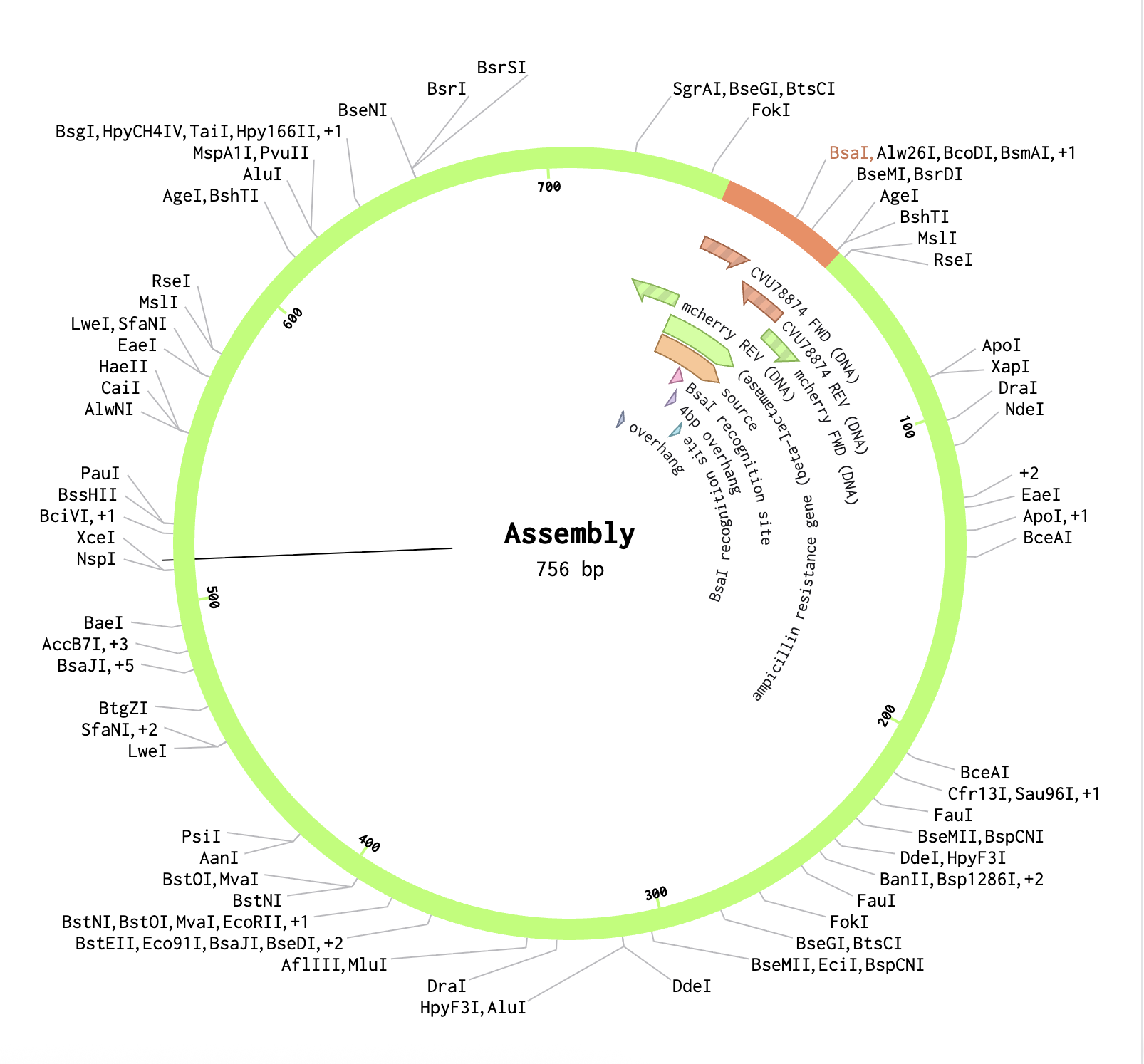
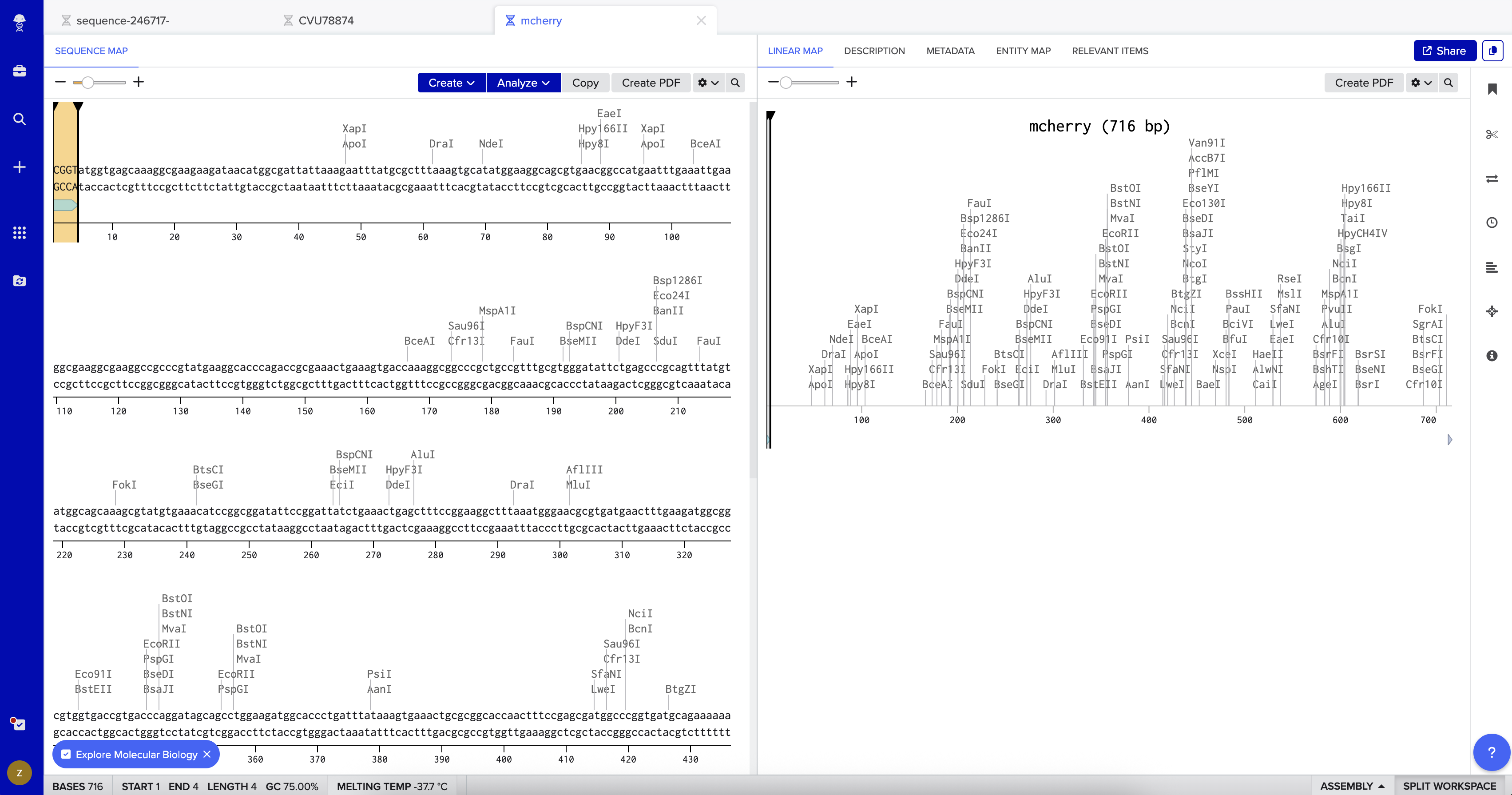
I then created a second DNA sequence (type: DNA, topology: linear) for the mCherry insert. From UniProt, I obtained the mCherry amino acid sequence of 236 amino acids and used the Reverse Translate tool at bioinformatics.org to convert it into a 708 bp DNA coding sequence optimized for E. coli. I added the BsaI-compatible overhangs to both ends of the sequence — CGGT at the 5’ end before the ATG start codon, and ACCG at the 3’ end after the stop codon — giving a final insert length of 716 bp.
In the pGEX sequence, I ran a digest using the Type IIS enzyme BsaI and found that it cuts once at position 2113 with 100% efficiency at 37°C. I added annotations for the cut site: 2107–2112 for the BsaI recognition site (GGTCTC), and 2113–2116 for the 4 bp overhang (CGGT) that gets exposed after cutting. Because BsaI cuts 1 nt outside its own recognition sequence, the recognition site is removed after digestion, leaving only the clean 4-nt sticky end for ligation.
Finally, using the Assembly Wizard, I selected Golden Gate Assembly as the method, assigned pGEX as the backbone and mCherry as the insert, and set the enzyme to BsaI. I manually selected the region around positions 2090–2130 as the fragment to provide sufficient flanking sequence for primer binding. The assembled plasmid was generated successfully, with mCherry correctly inserted at the BsaI cut site in the pGEX backbone, producing a circular plasmid where the GST tag and mCherry are expressed together as a fusion protein.