Individual Final Project
Secret Messengers: Steganography in the Genetic Code
SECTION 1: Abstract
The genetic code is degenerate, so that sixty-one sense codons specify only twenty amino acids, and most amino acid positions therefore admit between two and six synonymous codons that produce the same polypeptide. The project described here uses this redundancy as a small information-carrying channel within a functional, expressed coding sequence. A constraint-aware compiler takes a UTF-8 input message, applies Huffman compression, and maps the resulting bit stream onto the synonymous-codon choices at each amino acid position of either the superfolder variant of GFP (sfGFP; 238 amino acids; encoding capacity of 346 bits) or the red fluorescent protein mScarlet3 (229 amino acids; encoding capacity of 332 bits). A bit-rotation search then selects an offset that minimizes the longest homopolymer run and avoids internal NdeI and XhoI recognition sites within the insert. The resulting synthetic gene fragment is ordered from Twist Bioscience, double-digested with NdeI and XhoI, ligated into pET-28a(+), and transformed first into a high-efficiency cloning strain for propagation and subsequently into BL21(DE3) for T7-driven expression under IPTG induction. The fluorescence of the recombinant protein is then used as a first-pass quality check before any Sanger sequencing is performed: a colony that fails to glow under appropriate excitation indicates that the synonymous codon substitutions, despite passing all in silico checks, have nevertheless disrupted folding, maturation, or expression, and is filtered out, whereas a colony that does glow is progressed to Sanger sequencing for decoding through the browser encoder together with the stored metadata key. The working hypothesis is that the rotation search, together with random padding when zero padding fails to produce a clean rotation, will yield a Twist-synthesizable and pET-28a(+)-cloneable insert for the test messages, and that the resulting recombinant protein will glow in a comparable range to canonical-codon controls for the majority of constructs. The three specific aims examine round-trip fidelity of the full pipeline on a working test message, optimization of encoding modes along the two axes of capacity and stealth together with a curated fluorescent protein library, and the longer-term possibility of reading the encoded message directly from fluorescence patterns rather than from sequencing.
Demo and Presentation
Tool link: https://zbegum.github.io/htgaa-cipher.html/
SECTION 2: Project Aims
Aim 1: Communication Design and Wet Lab
The first aim of this project is to design, synthesize, clone, express, and decode a fluorescent-protein steganography insert carrying a hidden UTF-8 message within the coding sequence of either sfGFP or mScarlet3, by using Huffman compression of the input message, synonymous-codon wobble encoding of the resulting bit stream, bit-rotation optimization for synthesis and cloning constraints (all implemented in the encoder.py reference compiler with a parallel JavaScript browser implementation), Twist Bioscience gene fragment synthesis for the physical DNA construct, NdeI and XhoI restriction-ligation cloning into the pET-28a(+) expression vector, and a 96-well fluorescence assay in E. coli BL21(DE3) coupled to Sanger sequencing for decode verification.
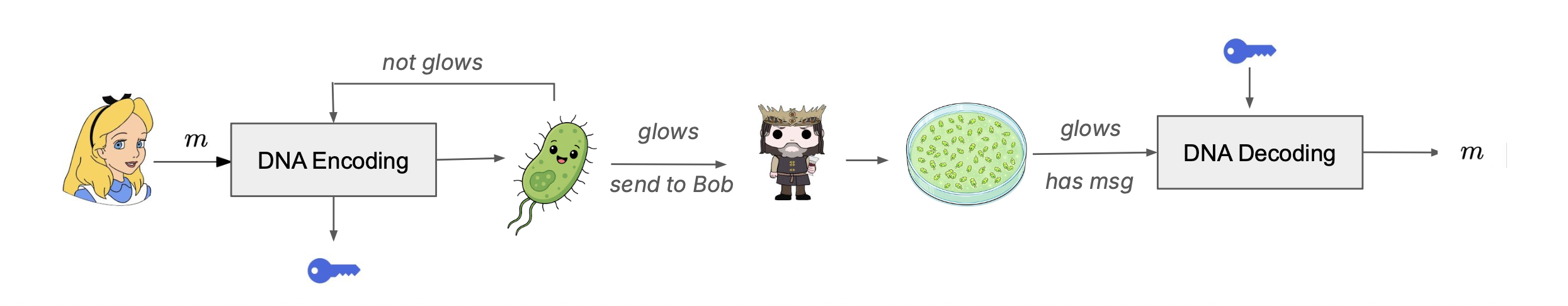
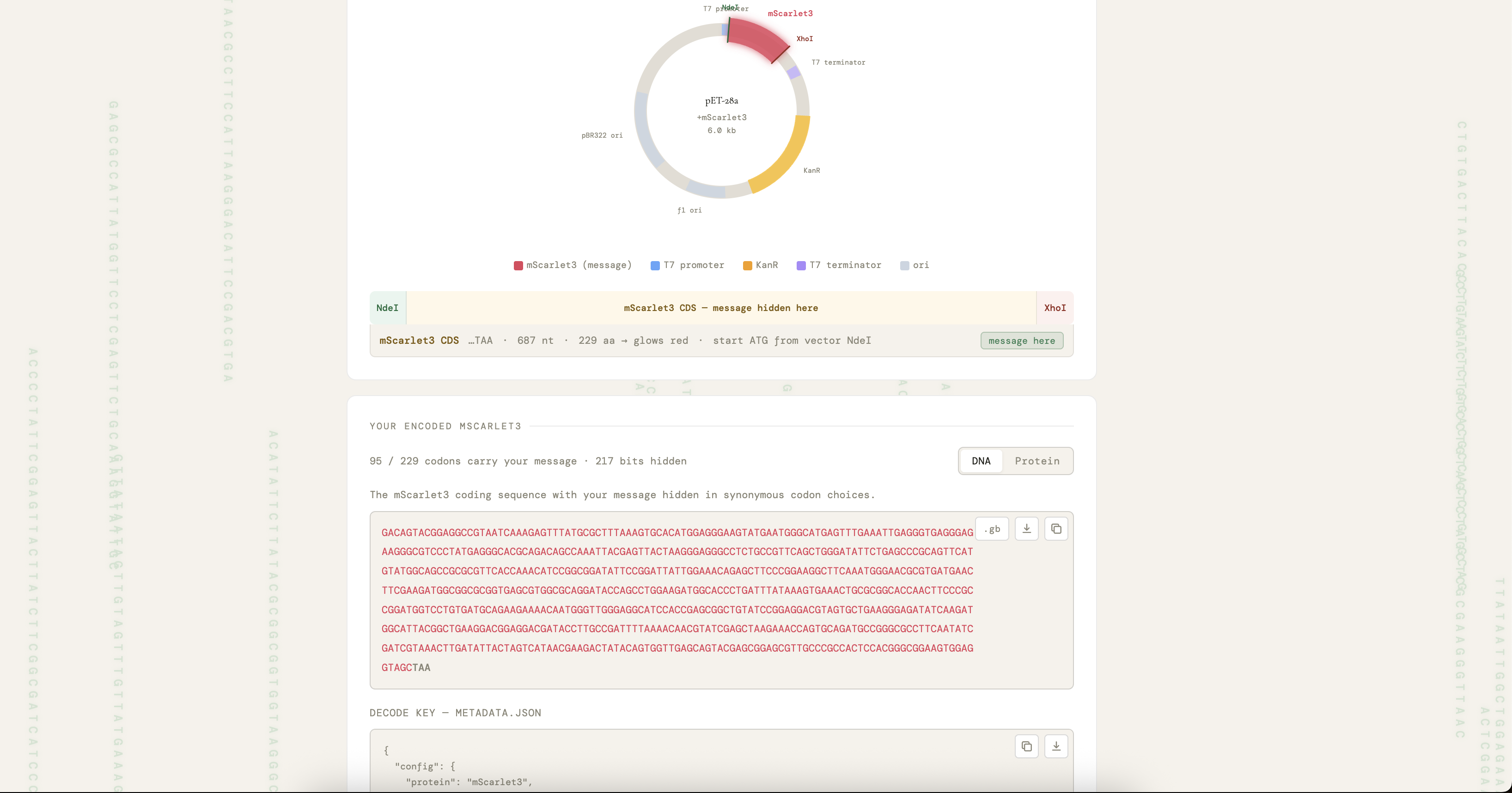
The DNA construct comprises a 687-nucleotide mScarlet3 insert encoding amino acid positions 2 through 229 plus a TAA stop codon (alternatively a 714-nucleotide sfGFP insert covering positions 2 through 238 plus a TAA stop codon), with the leading ATG start codon provided by the vector NdeI recognition sequence CATATG so that the synthesized fragment itself does not contain a start codon. The compiler enforces zero internal NdeI or XhoI recognition sites, a GC content between forty and sixty-five percent, and a maximum homopolymer run of six nucleotides, all of which lie within the documented manufacturability constraints of the Twist gene fragment synthesis service. The encode-side communication protocol is illustrated by the Alice-and-Bob diagram in the project presentation: Alice runs her message through the encoder, the encoder either accepts the rotation (good score) and returns a Twist-ready insert together with a metadata.json decoding key, or rejects it (bad score) and retries with random padding or a different rotation offset; Alice then orders the accepted insert from Twist as a clonal gene in pET-28a(+) and ships the resulting plasmid to Bob, who grows the colony, observes whether it glows, and on a positive fluorescence outcome proceeds to Sanger sequencing and decoding with the supplied key.
A Twist gene fragment order has been submitted for the mScarlet3 scaffold carrying the test message “Hello from HTGAA! We love growing almost anything.” The encoder reports 217 bits of payload occupancy out of the 332 total bits available in mScarlet3 (approximately sixty-five percent capacity utilization), an insert length of 687 nucleotides, a GC content of 52.7 percent, a longest homopolymer run of 4 nucleotides, a codon change rate relative to canonical of 41.5 percent, and a passing round-trip self-check confirming that the decoder reproduces the original message exactly from the emitted insert.
Aim 2: Encoding Optimization and Tool Improvements
The second aim of the project concerns extending the encoder along two axes that are in tension with one another, namely capacity and stealth. The capacity axis addresses how many more bytes can be encoded into a given fluorescent protein scaffold without increasing the insert length, and is bounded above by Σ⌊log₂(n)⌋ summed across all amino acid positions of the scaffold: for sfGFP the capacity is 346 bits, which after Huffman compression of English ASCII text corresponds to approximately seventy to eighty characters of payload, and for mScarlet3 the capacity is 332 bits, corresponding to approximately sixty-five to seventy-five characters. The stealth axis addresses whether the resulting coding sequence still looks like a plausible E. coli gene at the level of synonymous codon usage, since an encoder that always selects the rarest synonymous codon would maximize encoding capacity but produce a sequence with a low codon adaptation index relative to the E. coli K-12 baseline, which would be both detectable by a codon-deviation audit and potentially associated with reduced translation efficiency in vivo.
The medium-term development of the tool comprises two specific additions. The first is the support of multiple encoding modes, comparable side-by-side, so that the user can examine the trade-off between capacity, stealth (measured against the Kazusa K-12 codon profile), and the codon adaptation index for any given input message; candidate modes include the current greedy mode that prefers the most-favored codon when not constrained by the bit stream, a “stealth” mode that biases ties toward whichever choice preserves CAI most closely, and a “capacity” mode that maximizes the number of payload-carrying positions. The second is a curated fluorescent protein library that ships with the tool, comprising scaffolds that have been pre-validated for Twist printability and pET-28a(+) compatibility, and that allow the user to select among reporters with different capacities, spectral properties, and folding kinetics without having to reproduce the manual validation work that was required for the initial sfGFP and mScarlet3 scaffolds.
Aim 3: Talking DNAs
The third aim asks whether the Sanger sequencing step can be removed from the decoding workflow altogether, and the message read directly from the fluorescence output of the engineered bacteria. The inspiration for this direction comes from natural bioluminescent communication, in particular the patterned flashing of fireflies, which is itself a low-bandwidth optical signaling channel that has evolved over millions of years and which encodes species and mating information through timing and intensity rather than through any kind of sequence readout. If the engineered chassis carried more than one fluorescent reporter under independent regulatory control, the resulting strain could in principle express specific combinations of reporters in response to specific inducer conditions, and the visible fluorescence pattern across a series of induction cycles could itself function as a low-bandwidth communication channel, analogous to morse code in which short and long pulses encode the letters of a message (for instance, the morse encoding of “HELLO / HTGAA” reads “…. . .-.. .-.. — / …. - –. .- .-”).
The concrete proposal is a four-symbol language defined by a four-state machine, where each state corresponds to the expression of a distinct fluorescent reporter: state 1 is blue (TagBFP), state 2 is green (sfGFP), state 3 is red (mScarlet3), and state 4 is far-red (iRFP670). Each state therefore contributes two bits of information per visit, and a message is encoded as a directed walk through the state graph, with transitions driven by orthogonal small-molecule inducers such as anhydrotetracycline, IPTG, arabinose, and salicylate. A reader (whether a microscope, a plate reader, or in principle a phone camera with appropriate filters) records the colour visited at each induction cycle, and the resulting symbol sequence is decoded back to the original UTF-8 message. The morse encoding of “HELLO / HTGAA” shown above would, under this scheme, become a state sequence over the four-colour alphabet rather than a sequence of short and long pulses on a single channel.
Whether such a system could achieve a useful bandwidth without colour bleed-through between channels, whether the state transitions could be made reliable enough for the symbol error rate to compete with Sanger sequencing, whether the induction-cycle timing could be made fast enough for a meaningful end-to-end throughput, and whether the entire readout could be performed on consumer-grade imaging hardware rather than on a confocal microscope are all open questions that would have to be answered before Talking DNAs could realistically replace the Sanger-based decoding pipeline of Aim 1.
SECTION 3: Background
Literature Context
Two strands of prior work frame this project: information storage in DNA, and DNA watermarking. On the storage side, Church, Gao, and Kosuri (Science 337: 1628, 2012) encoded a 53,400-word book and several images directly into oligonucleotide pools and recovered the data, demonstrating that DNA can serve as a high-density archival medium. On the watermarking side, Heider and Barnekow (BMC Bioinformatics 8: 176, 2007) introduced the DNA-Crypt algorithm, which uses synonymous-codon substitutions within coding sequences to embed short identifying signatures and which uses the same per-position bit capacity formula adopted here; Clelland, Risca, and Bancroft (Nature 399: 533 to 534, 1999) had earlier shown in “Hiding messages in DNA microdots” that arbitrary text can be encoded in DNA bases and concealed within complex backgrounds. The fluorescent scaffold used here, superfolder GFP from Pédelacq and colleagues (Nature Biotechnology 24: 79 to 88, 2006), is a robust and widely used reporter for E. coli expression. This project borrows the codon-swapping trick from the watermarking line of work and uses it to carry arbitrary UTF-8 messages, while keeping the host gene functional and expressed, so that information storage and protein production occupy the same nucleotides rather than separate carrier DNA.
Innovation
The contribution of this project is small in scope but interesting in its framing. First, the encoder integrates Huffman compression, Kazusa-ordered synonymous-codon mapping, and a rotation search that avoids the NdeI and XhoI cut sites of the chosen cloning scheme, so that the resulting insert is directly orderable from Twist and directly cloneable into pET-28a(+) without manual edits. Second, the encode and decode pipelines run in two implementations, namely a JavaScript browser encoder and a Biopython-validated Python compiler, which share a deterministic codon ordering and produce byte-identical inserts from byte-identical inputs, so that the decoder can run on a separate machine using only the supplied metadata.json key. Third, and most speculatively, the in vivo fluorescence of the encoded reporter acts as a built-in receipt: a glowing colony tells the recipient that the DNA arrived intact enough to express, which is a necessary condition for the embedded message to be recoverable. Taken together, these three elements suggest that a single coding sequence may function as a small two-channel system carrying a protein and a message on the same nucleotides, and if the approach behaves as expected the same colony that glows on the petri dish also delivers an arbitrary digital payload to anyone holding the corresponding key.
Significance
The project is useful as a small synthetic biology exercise for a few related reasons. First, it gives a concrete demonstration that the redundancy of the genetic code can serve as an information channel inside an expressed coding sequence, while the resulting protein still folds and fluoresces under standard induction conditions in E. coli. Second, it turns codon usage bias into a one-experiment teaching example: changing the input message changes the DNA sequence, but the expressed protein is indistinguishable from canonical, which illustrates why synonymous substitutions are silent at the protein level but not at the nucleotide level. Third, the rotation search step is a small open-source tooling contribution that can be combined with existing codon adaptation index or codon-harmonization optimizers without major changes to those tools. Fourth, using fluorescence as a binary pre-filter before Sanger sequencing is a workflow improvement in any cloning setting where sequencing reactions are the rate-limiting cost or turnaround time, and the same idea generalizes to any cloning workflow that places a synonymous-codon-altered insert into a reporter scaffold. Finally, the project is small enough to complete within a course timeline while still touching most of the standard synthetic biology workflow, including DNA design, gene synthesis, restriction-based cloning verification, transformation, expression, microplate fluorescence assay, and Sanger sequencing.
Bioethical Considerations
Any technology that places digital data inside a functional, replicating biological agent raises dual-use considerations. The hidden channel could in principle carry sequences other than benign text, including toxin-gene fragments split across multiple synonymous positions, cryptographic credentials, or unauthorized watermarks identifying the originating laboratory. Even when the encoded payload is benign, the capability to embed data within engineered organisms shipped between laboratories raises modest questions about provenance and traceability, since recipient laboratories do not currently audit incoming strains for synonymous-codon anomalies as a matter of routine practice.
This project mitigates these concerns through several measures appropriate to a course context. First, the encoder source, codon table, and metadata format are open and documented, so any recipient can audit any construct produced by this pipeline against the vendor-default codon profile. Second, the host chassis is restricted to E. coli BL21(DE3), a lon⁻ and ompT⁻ B-strain not expected to survive outside laboratory media. Third, the encoder refuses payloads containing in-frame open reading frames, Shine-Dalgarno-consensus ribosome binding sites within the −4 to −12 window upstream of a downstream ATG, or known toxin-gene signatures from a curated deny-list. Fourth, Twist Bioscience applies its own biosecurity screen against the IGSC Harmonized Screening Protocol before synthesis. Finally, every plasmid emitted by the pipeline carries a provenance GenBank feature recording the encoder version, encoding date, and payload bit length, so downstream users are informed that the construct may carry an embedded message.
SECTION 4: Experimental Design
Workflow
The workflow is decomposed into seven steps. Step 1 runs on the laptop and produces a validated insert and a decode key. Step 2 outsources synthesis and cloning to Twist Bioscience. Steps 3 through 5 cover the wet-lab side, which reduces to transforming the delivered plasmid, inducing expression in 96-well format, and checking which colonies glow. Steps 6 and 7 cover decoding, which reduces to sequencing the glowing colonies and pasting the returned trace into the browser encoder. The fluorescence read at step 5 functions as a binary pre-filter for the sequencing step that follows, so non-glowing wells are excluded before any Sanger reaction is paid for. The decoder accepts the full plasmid Sanger trace directly through its NdeI-site anchored coding-sequence finder, so the user does not have to trim the trace down to the bare insert.
| # | Step | Method | Automation or instrument | Plate or tube | Expected result | Timeline |
|---|---|---|---|---|---|---|
| 01 | Encode and validate the insert | Run the encoder on the input UTF-8 payload to produce a Twist-ready insert: Huffman-compress the message, map the bit stream onto synonymous-codon positions in the host scaffold (sfGFP or mScarlet3) using the Kazusa K-12 ordering, run a bit-rotation search for zero internal NdeI or XhoI sites and a maximum homopolymer of four nucleotides, translate the open reading frame with NCBI table 1 to confirm the amino acid sequence matches the canonical reporter, then emit FASTA, GenBank, and the metadata.json decode key. | encoder.py and Bio.Restriction (laptop) | n/a | Round-trip-verified insert of 687 nucleotides (mScarlet3) or 714 nucleotides (sfGFP), with FASTA, GenBank, and metadata.json files. | Day 1, under one second |
| 02 | Order Twist clonal gene | Submit the validated insert as a Twist clonal gene in the pET-28a(+) context with NdeI and XhoI as the cloning sites; Twist performs the cloning and full sequence verification before shipping. | Twist Bioscience web portal | Twist clonal gene tube | Sequence-verified plasmid delivered approximately seven days later, with the vendor sequence report. | Days 2 to 9 |
| 03 | Transform into BL21(DE3) | Heat-shock 1 to 5 nanograms of the delivered plasmid into 50 microliters of BL21(DE3) chemically competent cells at 42 °C for thirty seconds, recover in SOC at 37 °C for one hour, and plate 100 microliters on LB plus kanamycin agar overnight at 37 °C. | Manual cold block with Eppendorf ThermoMixer C | Eppendorf 96-well deep-well plate, then Greiner 688161 LB plus Kan Petri dish | Discrete single transformant colonies the following morning. | Day 9 to 10, overnight |
| 04 | Set up and induce expression | Pick eight colonies per construct directly into a 96-well clear-bottom black plate prefilled with 200 microliters of LB plus kanamycin per well, grow at 37 °C to mid-log (OD600 of approximately 0.6), then induce with 0.5 mM IPTG and shift to 18 °C with shaking for eighteen hours. | Opentrons OT-2 transfer with BMG CLARIOstar Plus in growth mode | Corning 3603 96-well clear-bottom black plate | OD600 rises into the range of approximately 2 to 3 by the end of induction, with visible fluorescence in wells that express. | Days 10 to 11 |
| 05 | Glow check and pre-filter | Read end-point OD600 and end-point fluorescence at 485/510 nanometers (sfGFP) or 569/600 nanometers (mScarlet3), normalize fluorescence by OD600, and classify each well as glowing or non-glowing by thresholding against the empty-vector background at three standard deviations above the EV mean. | BMG CLARIOstar Plus microplate reader | Corning 3603 96-well clear-bottom black plate | List of glowing wells progressed to sequencing; non-glowing wells are flagged and excluded on the reasoning that the encoded reporter has failed to fold or mature. | Day 11, thirty minutes |
| 06 | Miniprep and Sanger sequence the glowing wells | Miniprep the cultures corresponding to glowing wells with the Zymo ZR Plasmid Miniprep Classic kit, then submit 500 nanograms of each miniprep with 25 picomoles of T7 promoter primer (sequence TAATACGACTCACTATAGGG) to a Sanger sequencing service. | Zymo 96-well plasmid kit with a Sanger sequencing service (Genewiz or Plasmidsaurus) | Zymo-Spin I-96 plate | Returned .ab1 electropherogram and base-call FASTA for each submitted plasmid. | Days 11 to 13 |
| 07 | Decode via browser encoder | Paste the returned Sanger FASTA directly into the browser encoder Decode tab together with the saved metadata.json key and run the decoder; the three-strategy CDS finder auto-anchors on the CATATG NdeI site, so no trimming of the trace is required, and the decoded message is compared against the original input. | Browser encoder Decode tab (htgaa-cipher.html) | n/a | Decoded message string matches the original input on the test cases that survived the glow filter; mismatches are localized by bit-diff against the Huffman-encoded original. | Day 13, instantaneous |
SECTION 5: Techniques, Tools, and Technology
The project exercises a standard set of synthetic biology techniques across the design, wet-lab, and verification stages:
- Design and validation: codon-aware coding-sequence engineering, restriction-site-aware design (zero internal NdeI or XhoI sites), and Biopython validation through
Bio.Seq,Bio.Restriction,Bio.SeqIO, andBio.SeqUtils.gc_fraction. - Synthesis and cloning: outsourced Twist clonal gene synthesis in the pET-28a(+) context.
- Expression: heat-shock transformation of E. coli BL21(DE3) and IPTG-induced T7-driven expression.
- Assay: 96-well microplate fluorescence on the BMG CLARIOstar Plus with Corning 3603 clear-bottom black plates.
- Verification: Zymo silica miniprep and outsourced Sanger sequencing through Genewiz or Plasmidsaurus.
- Automation: Opentrons OT-2 for transformation and culture-setup steps.
Three further techniques are deferred to later aims: multi-mode encoding with capacity, stealth, and CAI comparison and a curated fluorescent protein library are part of Aim 2; fluorescence-based readout in place of Sanger is the goal of Aim 3.
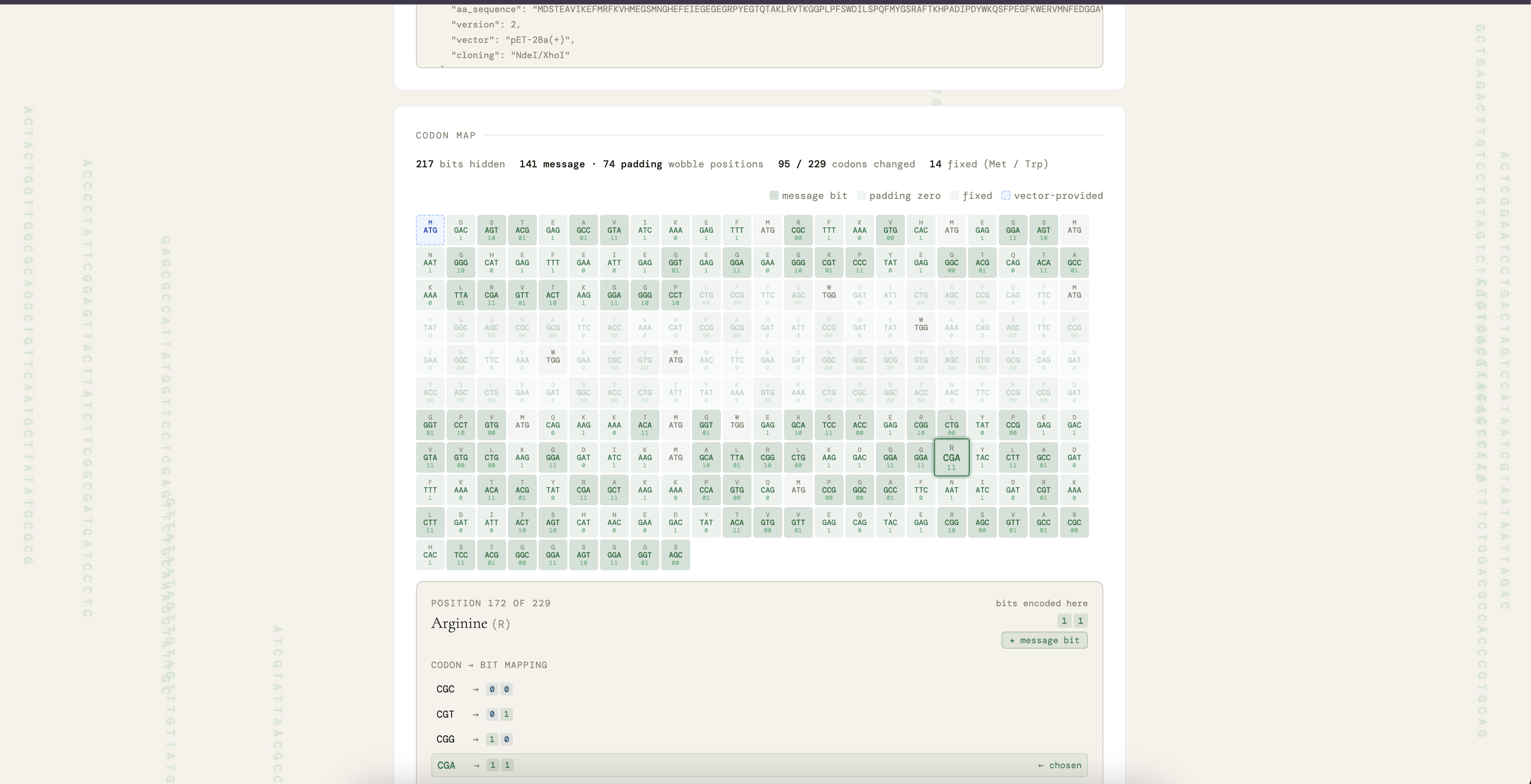
Two of these techniques are worth describing in more detail. The first is synonymous-codon wobble encoding, the core information-carrying mechanism of the project. The genetic code has 64 codons specifying only 20 amino acids and one stop, so 44 surplus codons are synonymous duplicates that can carry information without changing the encoded protein. At each amino acid position with n synonymous options, the encoder consumes ⌊log₂(n)⌋ bits from the Huffman-compressed message and uses them as an index into the codon list sorted by Kazusa K-12 frequency. The per-position contribution depends on the amino acid:
| Codons | Bits / position | Amino acids |
|---|---|---|
| 6 | 2 | Leu, Ser, Arg |
| 4 | 2 | Gly, Val, Ala, Thr, Pro |
| 3 | 1 | Ile (third codon unused) |
| 2 | 1 | Phe, Tyr, His, Gln, Asn, Lys, Asp, Glu, Cys |
| 1 | 0 | Met, Trp |
Summed across the 238 residues of sfGFP this gives 346 bits (approximately 70 to 80 Huffman-compressed characters), and the equivalent sum for the 229 residues of mScarlet3 is 332 bits (approximately 65 to 75 characters).
The second technique is cloning-aware bit rotation, the step that keeps the encoded insert directly orderable from Twist without manual edits. A naive encoding can place an NdeI site (CATATG) or an XhoI site (CTCGAG) inside the insert by chance, which would self-cleave during cloning. The encoder avoids this by trying cyclic rotations of the bit stream and scoring each by (internal cut count, longest homopolymer run):
The cut count dominates the lexicographic ordering, so any rotation with zero internal sites is preferred over any rotation that retains one. The chosen best_offset is recorded in metadata.json so the decoder can un-rotate the recovered bits before Huffman decoding. The procedure preserves both the encoding capacity and the amino acid sequence of the resulting protein, and runs in well under one second on either host scaffold.
SECTION 6: Project Validation
10a. Choice and rationale. The validation experiment is Sanger sequencing of each plasmid that passed the glow filter, with the returned trace fed through the browser encoder together with the saved metadata.json key. A successful round trip confirms in one step that the synthesized DNA matches the design, that the construct expresses, and that the encoder and decoder behave as inverse operations.
10b. Validation protocol.
- Pick wells that exceed the empty-vector background fluorescence by at least three standard deviations (step 5 of the workflow).
- Miniprep each selected well with the Zymo ZR kit and submit 500 ng of plasmid with 25 pmol of T7 promoter primer (TAATACGACTCACTATAGGG) to a Sanger service.
- Paste the returned base-call FASTA into the browser encoder Decode tab with
metadata.jsonand run the decoder. - Compare the decoded string against the original input; localize any mismatch by bit-diff against the Huffman-encoded original.
10c. Hypothetical data. A successful validation would show full bit recovery from the glowing wells and brightness within experimental noise of canonical:
| Construct | Bits encoded | Bits recovered | Brightness (WT = 1.00) |
|---|---|---|---|
| sfGFP, encoded | 256 | 256 | 0.98 ± 0.05 |
| mScarlet3, encoded (217-bit payload) | 217 | 217 | 1.01 ± 0.05 |
| EV (empty vector control) | n/a | n/a | 0.05 ± 0.01 |
Troubleshooting
The most important caveat is that the fluorescence pre-filter is necessary but not sufficient for successful decoding, since a glowing colony confirms only that the reporter still folds and matures, not that the codon string is free of point mutations; the mitigation is to keep Sanger sequencing as the authoritative validation step rather than to rely on glow alone. A second concern is the capacity ceiling: the 346-bit capacity of sfGFP corresponds to approximately seventy to eighty Huffman-compressed characters, which is enough for a short message but not for a paragraph, and the alternative is to migrate to a larger host protein such as firefly luciferase (550 residues, approximately 850 bits) or to chain reporters within a P2A polyprotein cassette, as outlined in Aim 2.
SECTION 7: Additional Information
References
- Pédelacq, J.-D., Cabantous, S., Tran, T., Terwilliger, T. C., and Waldo, G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nature Biotechnology 24: 79 to 88. doi:10.1038/nbt1172
- Gadella, T. W. J., et al. (2023). mScarlet3: a brilliant and fast-maturing red fluorescent protein. Nature Methods 20: 541 to 545. doi:10.1038/s41592-023-01809-y
- Clelland, C. T., Risca, V., and Bancroft, C. (1999). Hiding messages in DNA microdots. Nature 399: 533 to 534. doi:10.1038/21092
- Heider, D., and Barnekow, A. (2007). DNA-based watermarks using the DNA-Crypt algorithm. BMC Bioinformatics 8: 176. doi:10.1186/1471-2105-8-176
- Cock, P. J. A., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25(11): 1422 to 1423. doi:10.1093/bioinformatics/btp163
- Huffman, D. A. (1952). A method for the construction of minimum-redundancy codes. Proceedings of the IRE 40(9): 1098 to 1101.
- Kazusa Codon Usage Database, Escherichia coli K-12 (NCBI taxonomy identifier 83333): https://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=83333
- pET-28a(+) expression vector, Novagen / MilliporeSigma; Addgene plasmid 26094: https://www.addgene.org/26094/
- FPbase entry for mScarlet3: https://www.fpbase.org/protein/mscarlet3/
- FPbase entry for superfolder GFP: https://www.fpbase.org/protein/superfolder-gfp/
Supplies and Budget
The principal out-of-pocket cost is the Twist clonal gene order in the pET-28a(+) context at approximately $120 per construct, plus Sanger sequencing through Genewiz or Plasmidsaurus. All other reagents and consumables (BL21(DE3) competent cells, kanamycin, IPTG, Zymo miniprep kit, Corning 3603 black plates, LB media) are part of the standard course inventory and are not separately budgeted here.