Week 4: Protein Design Part I
Part A: Conceptual Questions
Q1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
500g of meat contains approximately 125g of protein. One mole of a 100-Dalton molecule weighs 100 grams and Avogadro’s number (6.022 × 10²³) is the number of molecules in one mole. So 125g of protein ÷ 100 g/mol = 1.25 moles of amino acid residues, multiplied by Avogadro’s number: 1.25 × 6.022 × 10²³ ≈ 7.5 × 10²³ amino acid molecules.
Q2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Digestive enzymes(proteases like pepsin and trypsin) break down the cow’s proteins into individual amino acids via hydrolysis. They are absorbed into our bloodstream and our cells reassemble them into human proteins following the instructions in DNA.
Q3. Why are there only 20 natural amino acids?
The 20 amino acids cover a broad range of chemical properties, positive and negative charges, hydrophobic and hydrophilic, large and small, rigid and flexible structures. This set is chemically diverse enough to build proteins capable of catalyzing thousands of reactions and forming diverse structures. The triplet codon system (64 codons from 4 bases) naturally accommodates around 20 amino acids after accounting for stop codons and redundancy. Early life developed and evolved from this set.
Q5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids form spontaneously through non-biological chemistry. The Miller-Urey experiment (1953) demonstrated that sparking a mixture of water, methane, ammonia, and hydrogen(simulating early Earth conditions) produced multiple amino acids without any enzymes or cells. Amino acids have also been found on meteorites and detected in interstellar molecular clouds. Energy sources like lightning, UV radiation, and hydrothermal vents can drive the formation of amino acids from simple precursors such as hydrogen cyanide and ammonia. Amino acids are not uniquely biological but are basic organic chemistry that life later organized into a precise system.
Q6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
It would form a left-handed α-helix. Natural L-amino acids produce right-handed α-helices because the L-configuration creates specific steric preferences in backbone angles that favor a right-handed spiral. D-amino acids are the mirror of L-amino acids, so preferences flip and the resulting helix spirals in the opposite direction.
Q7. Can you discover additional helices in proteins?
Yes. Beyond the standard α-helix, other helical forms exist. The 3₁₀ helix is tighter, with hydrogen bonds between amino acid i and i+3 (compared to i and i+4 in the α-helix), making it narrower. The π-helix is wider and looser, with i to i+5 hydrogen bonds. Polyproline helices (type I and II) have no internal hydrogen bonds.
Q8. Why are most molecular helices right-handed?
Life and protein-making exclusively uses L-amino acids, and the L-configuration creates steric preferences that favor right-handed helices. Specifically, in an L-amino acid, the side chain clashes with the backbone carbonyl in a left-handed spiral, which is energetically unfavorable.
Q9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets are extended, flat structures with hydrogen bond donors and acceptors exposed along their edges. These unsatisfied hydrogen bonding sites in their edges will bond to those of other β-sheets. This is inherently self-propagating because the outward facing edges drive intermolecular aggregation. The flat faces of stacked β-sheets also interact through hydrophobic packing forces.
Q10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloid diseases involve proteins that misfold into β-sheet-rich structures. The β-sheet conformation is a generic, low-energy state that doesn’t require the precise side-chain packing of a native fold. When a protein misfolds and exposes β-sheet edges, it templates neighboring proteins to misfold the same way, creating a self-propagating cascade. Amyloid β-sheets can be used as materials because they have tensile strength comparable to steel, resist heat and proteases, and self-assemble spontaneously. Researchers have used them as scaffolds for tissue engineering, conductive nanowires, adhesive coatings, and hydrogel matrices for cell culture.
Part B: Protein Analysis and Visualization
Protein Selection
I selected human heavy-chain ferritin (HuHF), the primary iron-storage protein found in virtually all living organisms. I chose this because its self-assembling architecture makes it a powerful platform for nanotechnology applications including drug delivery, biosensor scaffolding, and templated nanomaterial synthesis. Ferritin represents a natural example of the design principle: simple modular subunits that autonomously organize into functional nanostructures.
Sequence Analysis
MTTASTSQVRQNYHQDSEAAINRQINLELYASYVYLSMSYYFDRDDVALKNFAKYFLHQSHEEREHAEKLMKLQNQRGGRIFLQDIKKPDCDDWESGLNAMECALHLEKNVNQSLLELHKLATDKNDPHLCDFIETHYLNEQVKAIKELGDHVTNLRKMGAPESGLAEYLFDKHTLGDSDNESThe protein is 183 amino acids long. The most frequent amino acid is leucine (L).
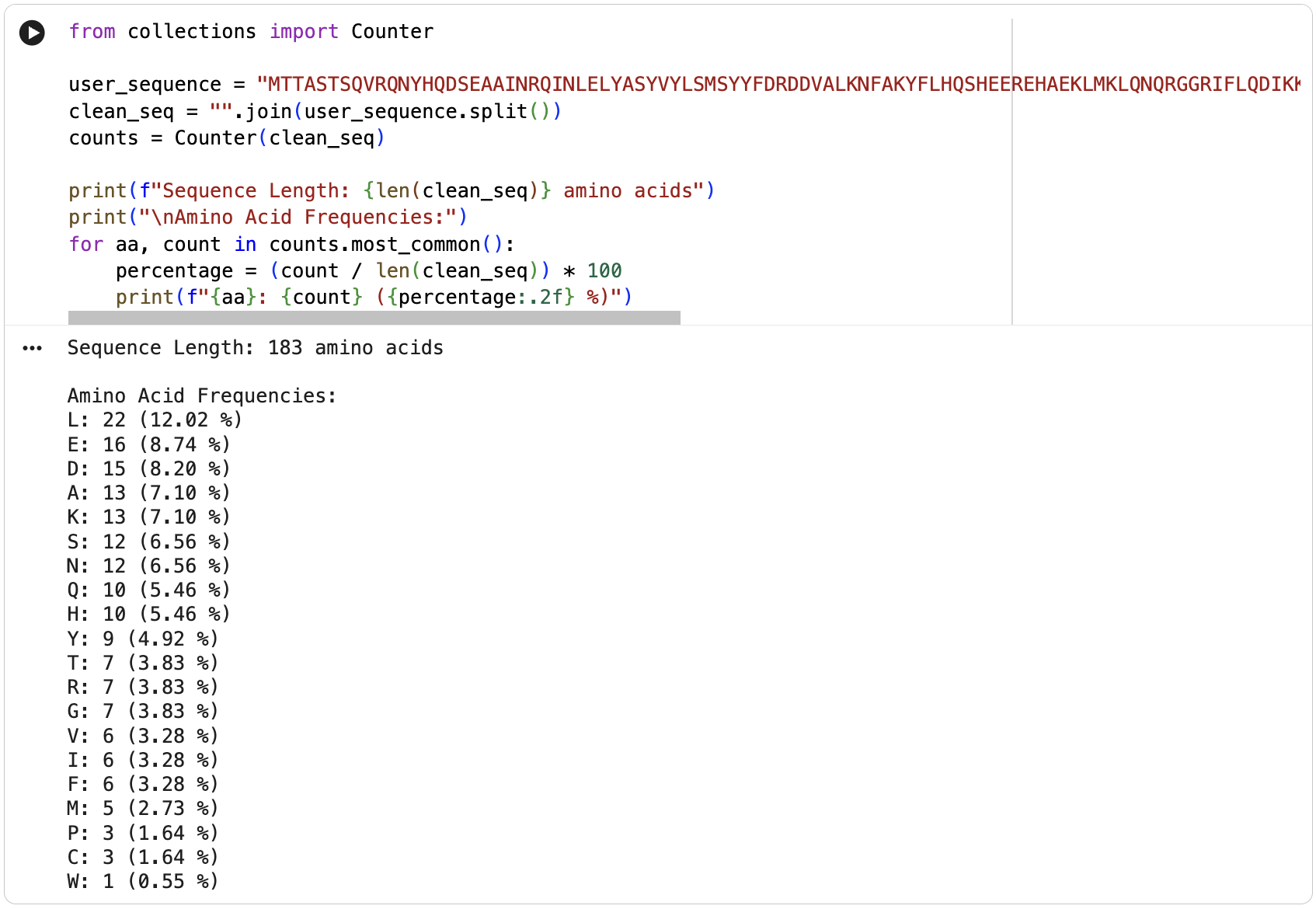
Homologs: BLAST search of UniProt identified at least 250 homologs at ≥90% sequence identity, including primates, other mammals, and even bacteria.
Protein Family: Ferritin belongs to the ferritin-like superfamily (Pfam: PF00210). This superfamily also includes bacterioferritins and DNA-binding proteins from starved cells (Dps proteins). InterPro classifies it under the ferritin/ribonucleotide reductase-like domain family.
RCSB Structure
Protein Structure on RCSB: https://doi.org/10.2210/pdb1FHA/pdb
When was it solved: deposited in 1990 and released in 1992, with the primary citation published in 1991 (Lawson et al., Nature 349: 541-544). It was solved by X-ray crystallography at a resolution of 2.40 Å(good quality). The R-value of 0.205 further confirms the model fits the experimental data well.
Other molecules in the structure: The structure contains two types of non-protein molecules: iron (Fe³⁺) ions located at the ferroxidase center within each of its four-helix bundle, which is where ferritin catalyzes the oxidation of Fe²⁺ to Fe³⁺ for storage; and calcium (Ca²⁺) ions, likely present from crystallization conditions.
Structure classification: The protein is classified as a METAL BINDING PROTEIN with the enzyme classification EC 1.16.3.1 (ferroxidase). The biological assembly has octahedral symmetry (O) as a homo 24-mer (A24), with each subunit folded as a four-helix bundle.
PyMOL Visualization
 Cartoon visualization
Cartoon visualization
 Ribbon visualization
Ribbon visualization
 Ball and stick visualization
Ball and stick visualization
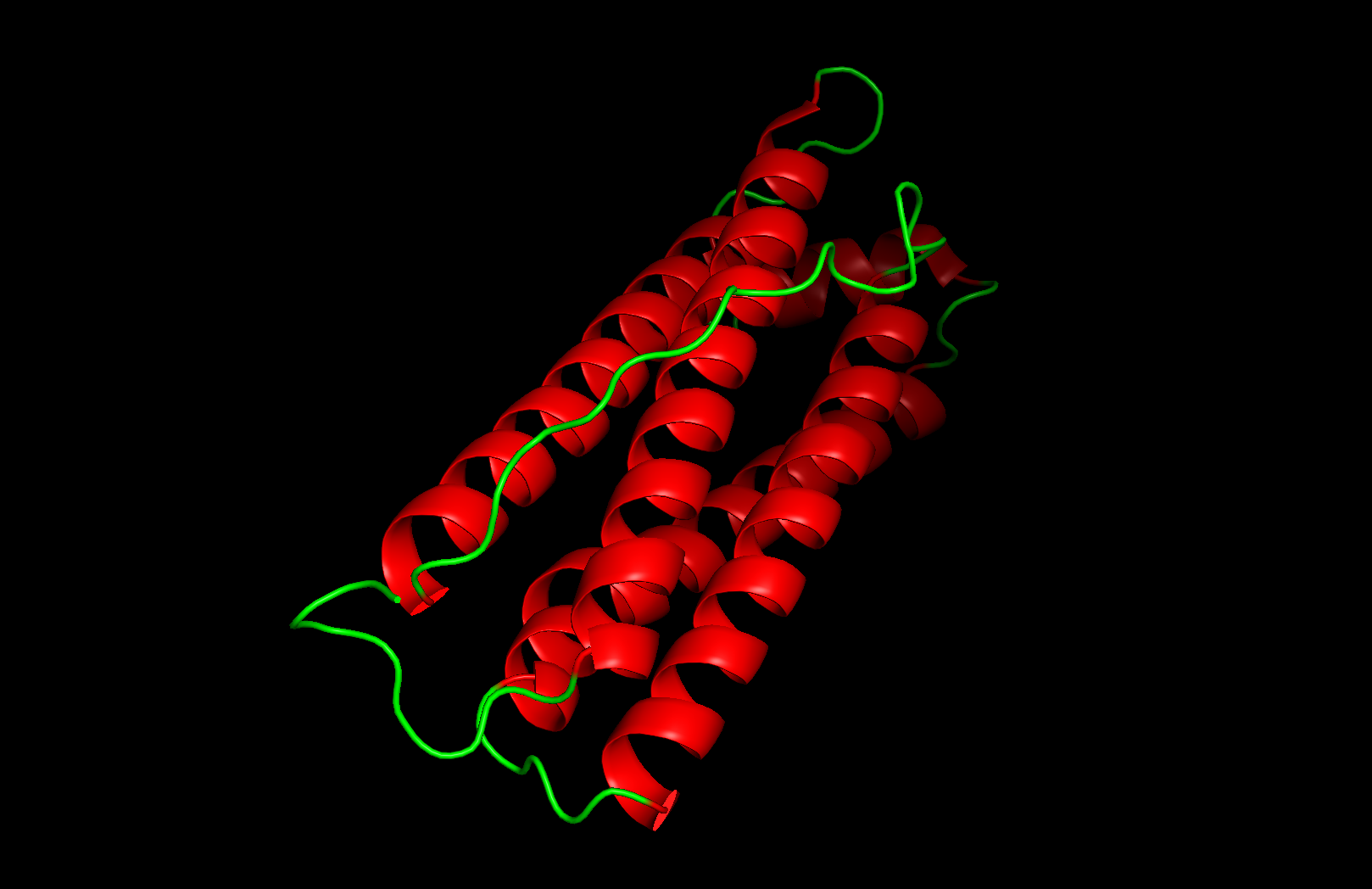 Secondary structure: The protein is almost entirely alpha-helical (red) with no beta-sheets. The only non-helical regions (green) are short loops connecting the four main helices.
Secondary structure: The protein is almost entirely alpha-helical (red) with no beta-sheets. The only non-helical regions (green) are short loops connecting the four main helices.
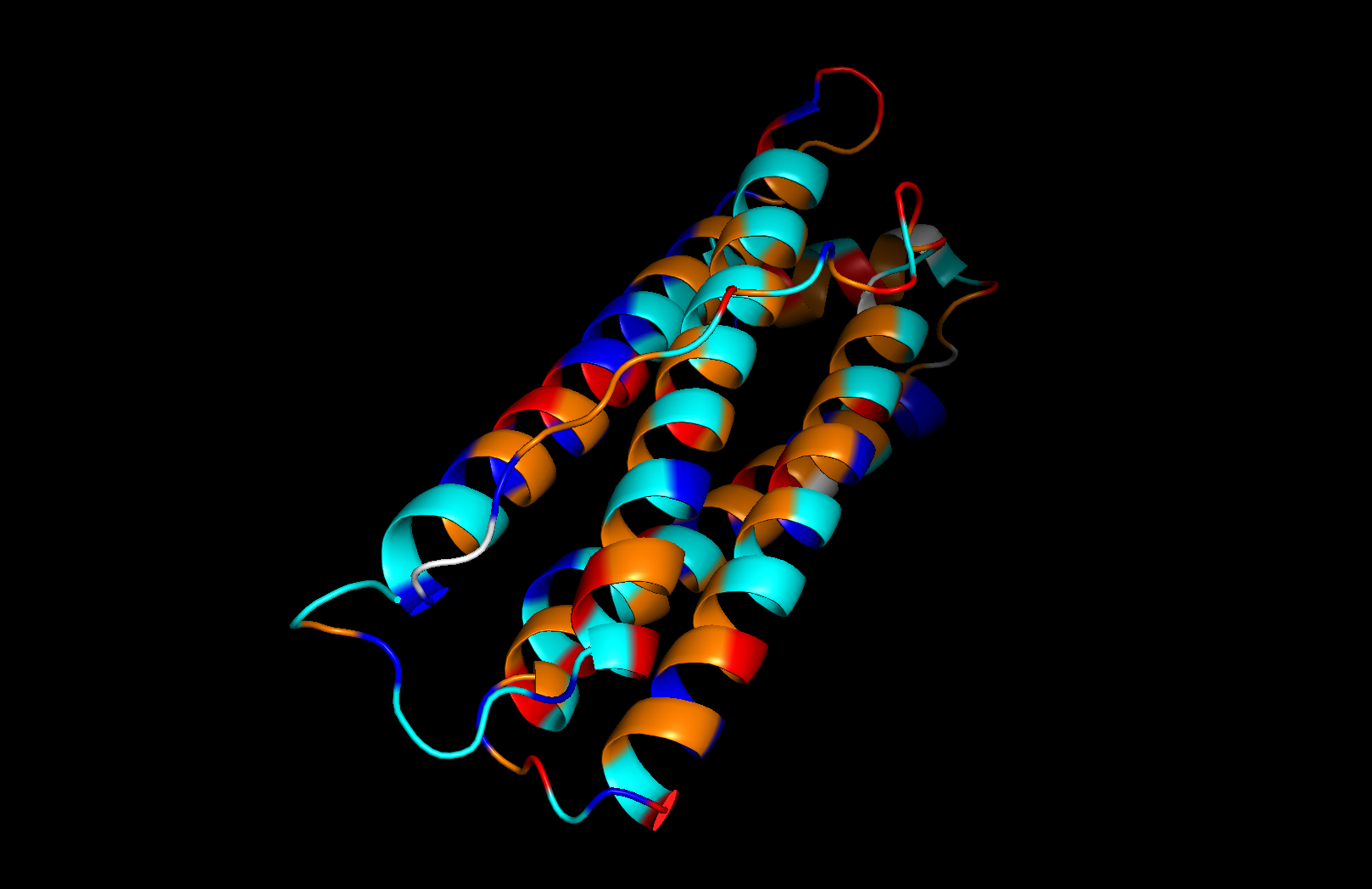 Residue type: Hydrophobic residues (orange) and hydrophilic/charged residues (cyan, blue, red) are interspersed along the helices, but the hydrophobic residues face inward toward the bundle core while charged and polar residues face outward toward solvent. This inside-out arrangement is typical of soluble proteins.
Residue type: Hydrophobic residues (orange) and hydrophilic/charged residues (cyan, blue, red) are interspersed along the helices, but the hydrophobic residues face inward toward the bundle core while charged and polar residues face outward toward solvent. This inside-out arrangement is typical of soluble proteins.
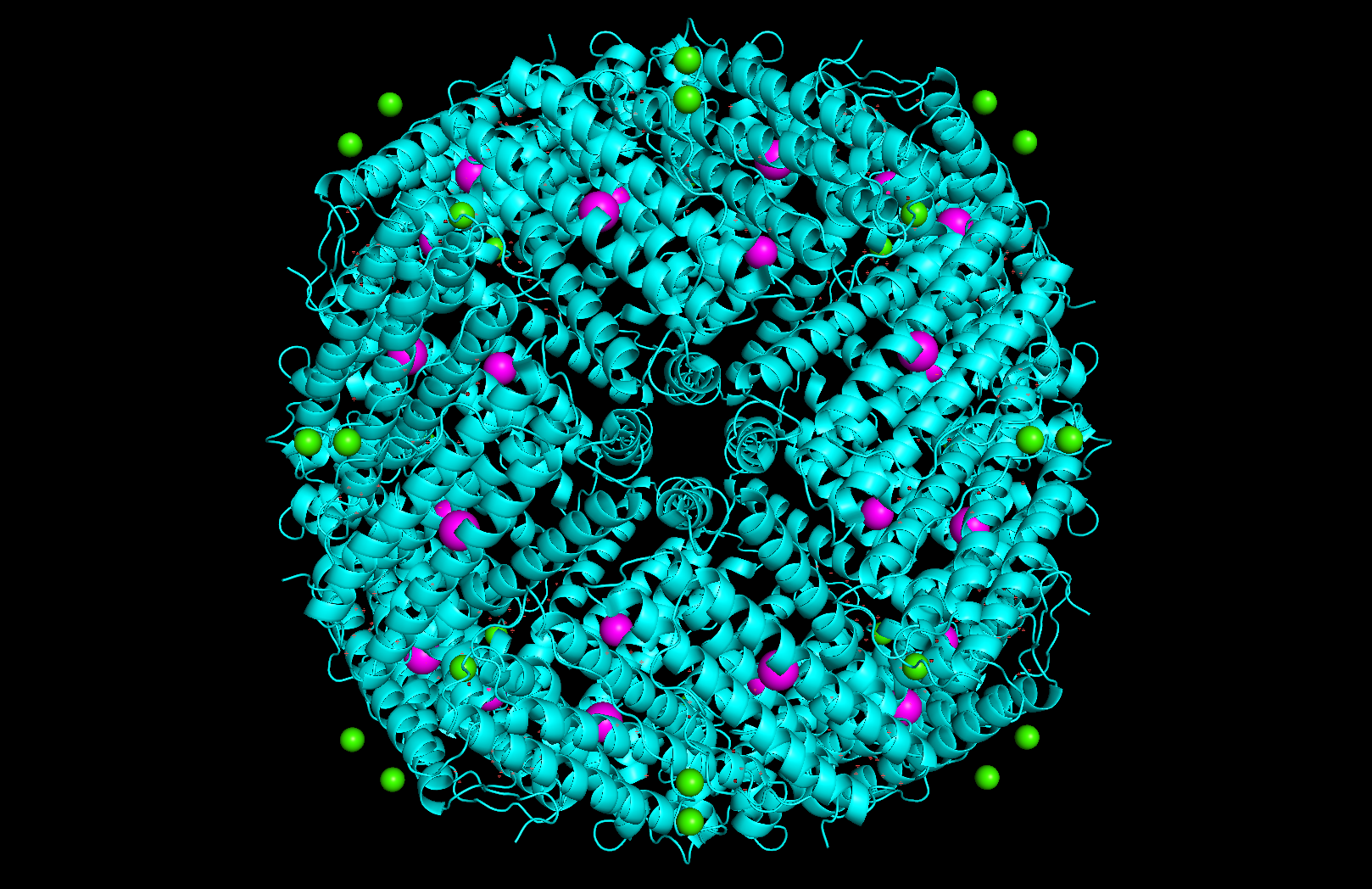 Surface and binding pockets: The 24-mer assembly reveals a hollow spherical cage with visible pores which are the channels through which iron ions enter and exit the interior cavity. Iron atoms (magenta spheres) are visible at the ferroxidase centers within each subunit, and calcium ions (green spheres) sit at the symmetry axes.
Surface and binding pockets: The 24-mer assembly reveals a hollow spherical cage with visible pores which are the channels through which iron ions enter and exit the interior cavity. Iron atoms (magenta spheres) are visible at the ferroxidase centers within each subunit, and calcium ions (green spheres) sit at the symmetry axes.
Part C: Using ML-Based Protein Design Tools
For Part C, I chose Reflectin A1 from Doryteuthis pealeii (longfin inshore squid), the protein responsible for dynamic structural coloration in cephalopod skin.
MNRYLNRQRLYNMYRNKYRGVMEPMSRMTMDFQGRYMDSQGRMVDPRYYDYYGRMHDHDRYYGRSMFNQGHSMDSQRYGGWMDNPERYMDMSGYQMDMQGRWMDAQGRFNNPFGQMWHGRQGHYPGYMSSHSMYGRNMYNPYHSHYASRHFDSPERWMDMSGYQMDMQGRWMDNYGRYVNPFNHHMYGRNMCYPYGNHYYNRHMEHPERYMDMSGYQMDMQGRWMDTHGRHCNPFGQMWHNRHGYYPGHPHGRNMFQPERWMDMSGYQMDMQGRWMDNYGRYVNPFSHNYGRHMNYPGGHYNYHHGRYMNHPERHMDMSSYQMDMHGRWMDNQGRYIDNFDRNYYDYHMYC1. Protein Language Modeling
Deep Mutational Scan
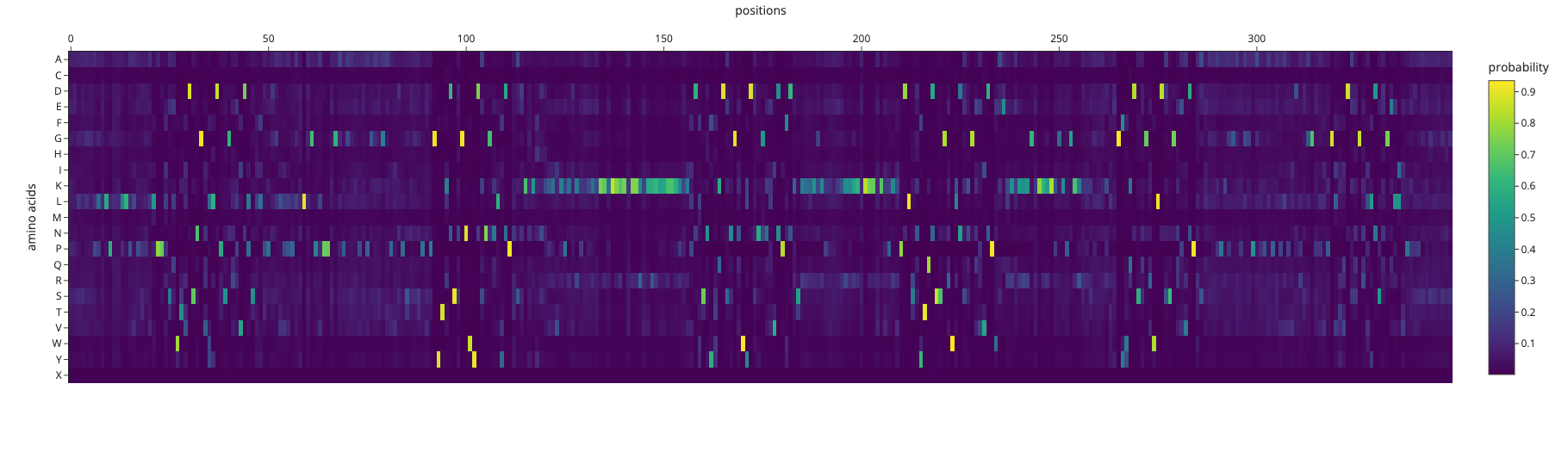
I used ESM-2 to perform the deep mutational scan of Reflectin A1 (350 aa) in RELATIVE mode, scoring the predicted effect of every possible single amino acid substitution at every position.
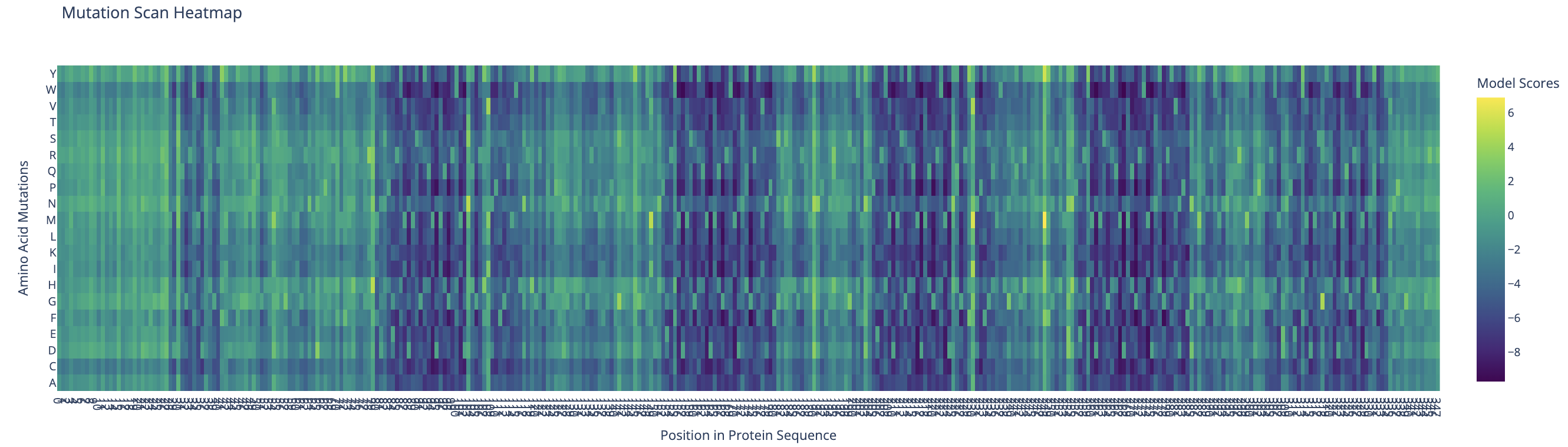 ESM-2 deep mutational scan heatmap. X-axis: position in sequence (1–350). Y-axis: amino acid substitutions. Color: model score (yellow = tolerated, dark blue/purple = deleterious).
ESM-2 deep mutational scan heatmap. X-axis: position in sequence (1–350). Y-axis: amino acid substitutions. Color: model score (yellow = tolerated, dark blue/purple = deleterious).
The heatmap reveals regularly spaced clusters of mutation-intolerant positions approximately every 50–70 residues. This periodicity suggests the model has learned the repeating internal motif structure of the reflectin family. Cysteine(C) and tryptophan(W) substitutions are the most strongly penalized across these positions, likely because introducing disulfide-forming or large aromatic residues at these sites would disrupt the native fold.
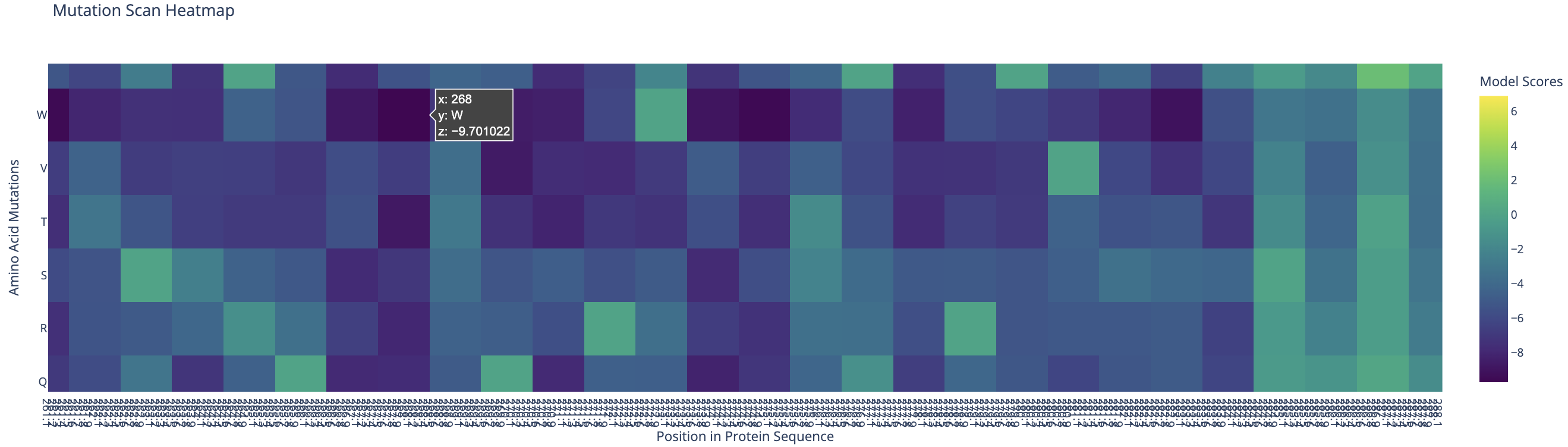
Standout residue: Position 268 mutated to tryptophan (W) scored −9.70, the most deleterious prediction in the scan. It is also intolerant to methionine, leucine, lysine, and isoleucine, which suggests the original residue at this position is structurally or functionally critical and cannot be replaced without disrupting the protein.
Latent Space Analysis
I embedded 15,177 proteins from the ASTRAL SCOP structural classification database using ESM-2 mean token embeddings (320 dimensions), then reduced them to 3D with t-SNE for visualization. The resulting plot shows clear clustering — proteins with similar sequences and structures group into distinct neighborhoods, confirming that ESM-2’s learned representations capture meaningful structural relationships.
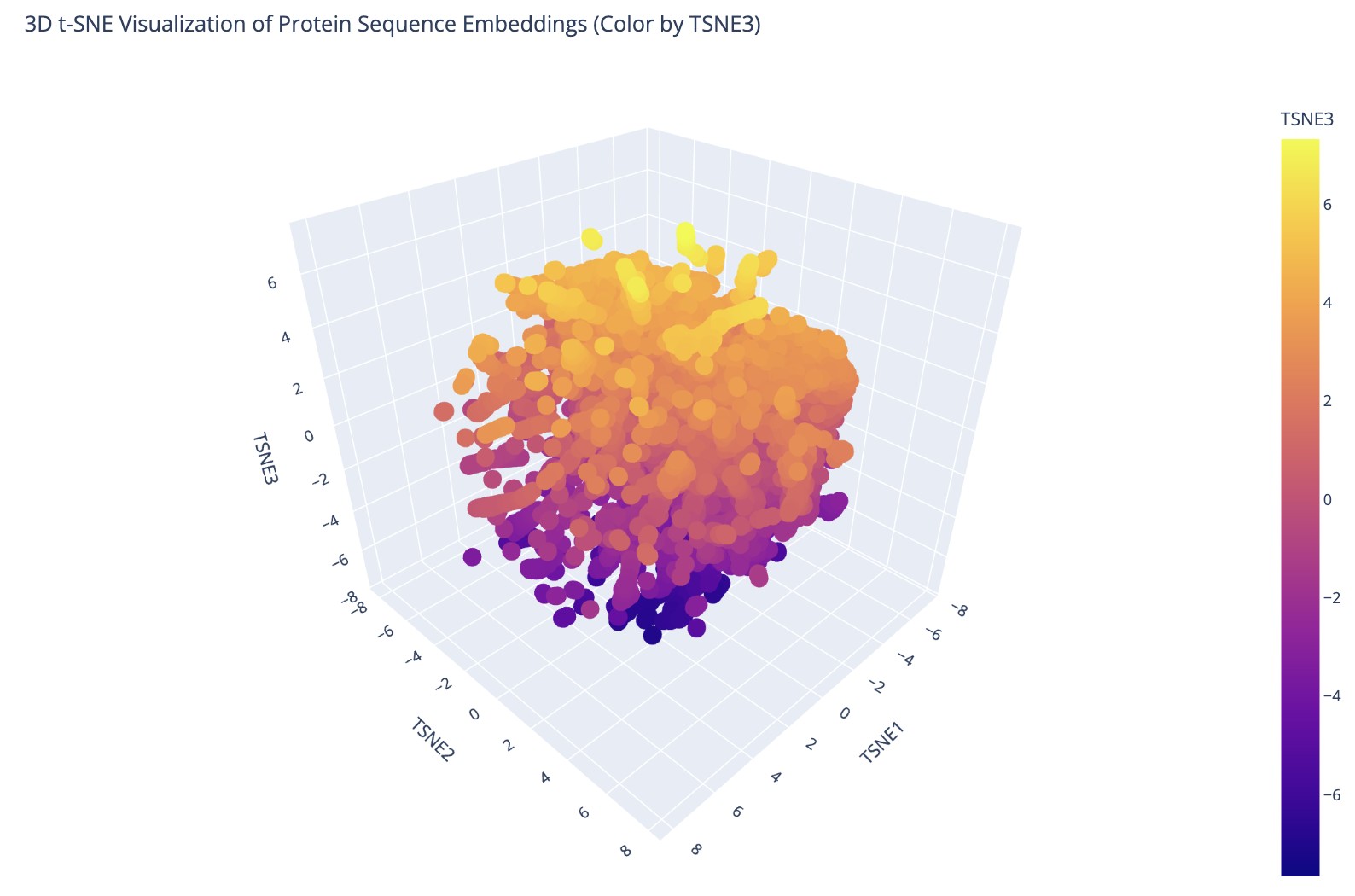 3D t-SNE visualization of 15,177 ASTRAL SCOP protein embeddings colored by TSNE3 component. Distinct clusters correspond to different protein fold families.
3D t-SNE visualization of 15,177 ASTRAL SCOP protein embeddings colored by TSNE3 component. Distinct clusters correspond to different protein fold families.
I then embedded Reflectin A1 into the same space and projected it alongside the ASTRAL dataset.
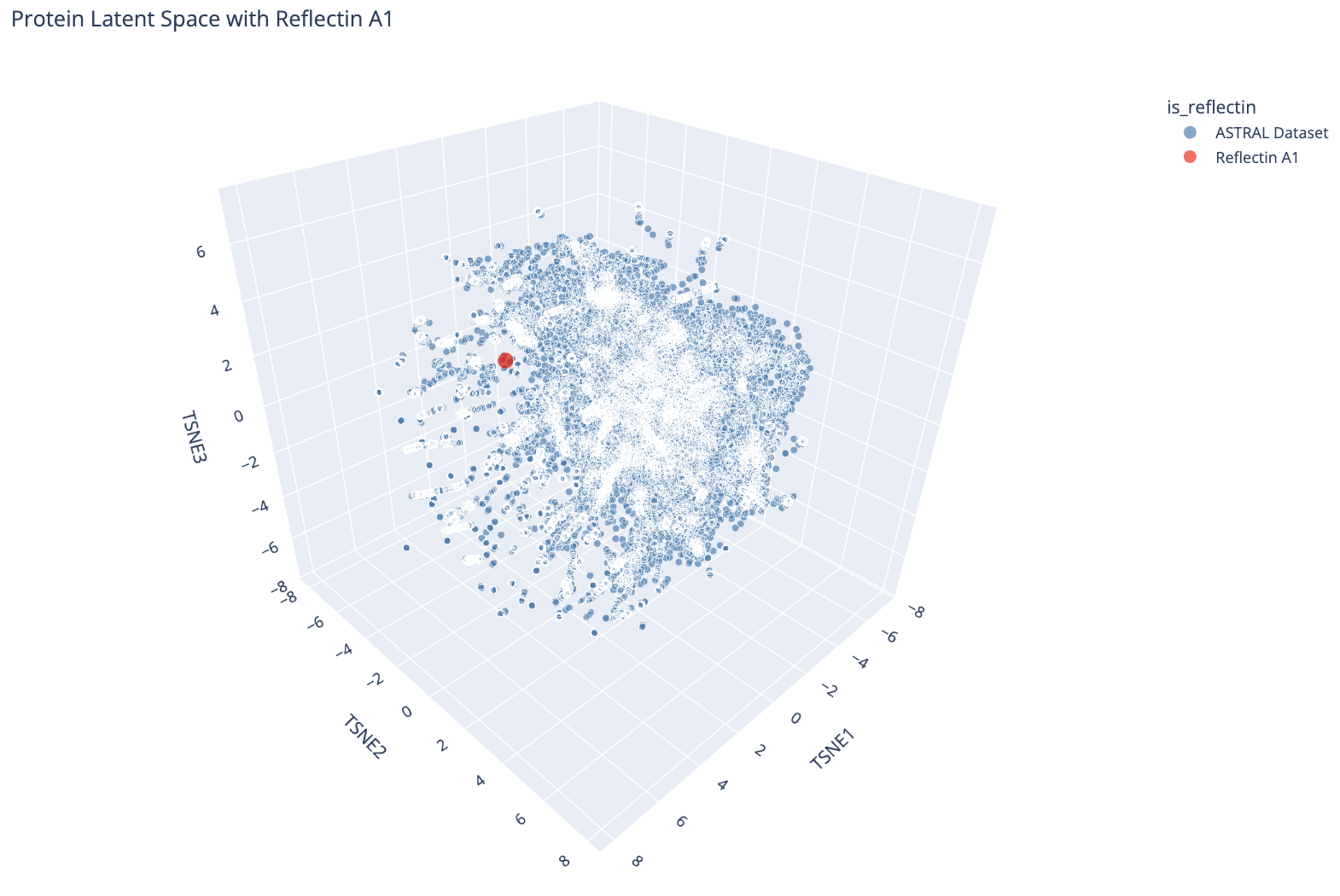 Reflectin A1 (red dot) plotted in the ESM-2 latent space alongside the ASTRAL dataset (blue). It sits near the periphery of the main cluster.
Reflectin A1 (red dot) plotted in the ESM-2 latent space alongside the ASTRAL dataset (blue). It sits near the periphery of the main cluster.
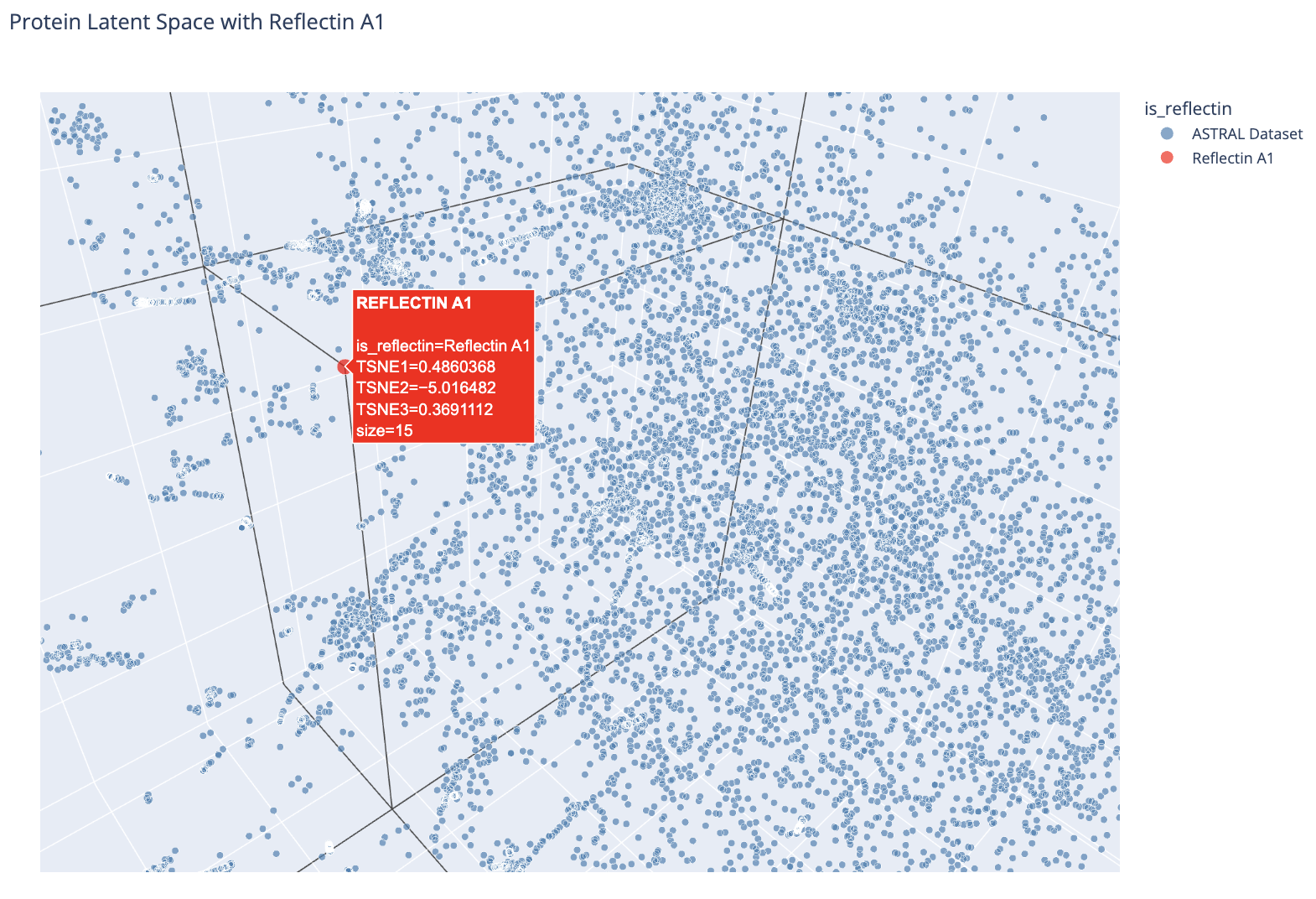 Zoomed view showing Reflectin A1’s position relative to nearby ASTRAL proteins.
Zoomed view showing Reflectin A1’s position relative to nearby ASTRAL proteins.

Using Euclidean distance in the original 320-dimensional embedding space, the 5 nearest neighbors to Reflectin A1 are:
- Tachylectin-2, Japanese horseshoe crab (dist: 3.81)
- Rabbit protein, automated match (dist: 3.87)
- Hemolytic lectin CEL-III, sea cucumber (dist: 4.09)
- Methionine-rich 2S albumin, sunflower (dist: 4.25)
- Clostridium thermocellum protein, automated match (dist: 4.27)
Two of the five closest neighbors are lectins with repetitive structural motifs used for carbohydrate binding. Reflectin also contains repetitive internal motifs, so ESM-2 may be picking up on this shared repetitive sequence character despite the proteins being functionally unrelated. The methionine-rich neighbor is also notable since reflectin is unusually rich in methionine. The relatively large distances across all five neighbors (3.8–4.3) confirm that reflectin has no close structural homologs in the database, consistent with it being a structurally novel protein unique to cephalopods.
C2. Protein Folding
Folding with ESMFold
I folded Reflectin A1 using ESMFold and compared the result to the AlphaFold prediction.
| pTM | pLDDT | |
|---|---|---|
| Original | 0.157 | 39.15 |
ESMFold produced very low confidence scores: a pLDDT of 39.15 (out of 100) and pTM of 0.157 (out of 1). Values below 50 pLDDT indicate intrinsic disorder. The structure is almost entirely red when colored by confidence, meaning ESMFold cannot confidently predict any region of the fold.
 ESMFold prediction colored by pLDDT confidence. Red = very low confidence, yellow = slightly higher confidence. The protein is almost entirely red.
ESMFold prediction colored by pLDDT confidence. Red = very low confidence, yellow = slightly higher confidence. The protein is almost entirely red.
 AlphaFold prediction for comparison. Both models show the protein is largely disordered, but AlphaFold identifies a few small β-sheet domains (blue) within the repetitive regions that ESMFold misses entirely.
AlphaFold prediction for comparison. Both models show the protein is largely disordered, but AlphaFold identifies a few small β-sheet domains (blue) within the repetitive regions that ESMFold misses entirely.
Mutation Resilience
I tested whether the predicted structure is sensitive to mutations by single substitutions and large segment replacements:
| Variant | Mutation | pTM | pLDDT |
|---|---|---|---|
| Original | — | 0.157 | 39.15 |
| Q268W | Single residue (position 268) | 0.157 | 39.59 |
| W81A | Single residue (position 81) | 0.157 | 39.52 |
| Ala segment | Positions 100–130 → all alanine | 0.152 | 38.60 |
| Gly segment | Positions 200–230 → all glycine | 0.153 | 36.48 |
 Q268W single mutation: virtually identical to original.
Q268W single mutation: virtually identical to original.
 W81A single mutation: virtually identical to original.
W81A single mutation: virtually identical to original.
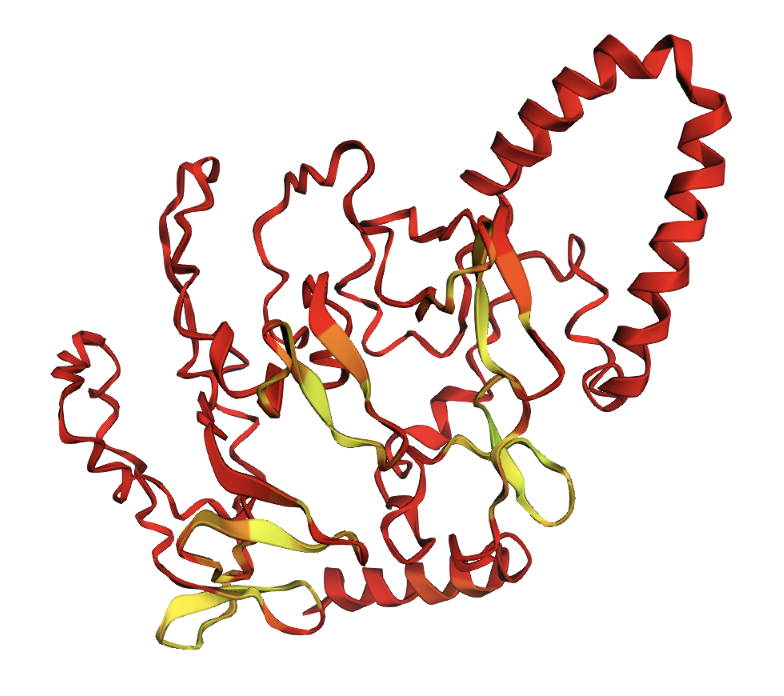 Positions 100–130 replaced with all alanines. A helix appears in the alanine region, but confidence remains low.
Positions 100–130 replaced with all alanines. A helix appears in the alanine region, but confidence remains low.
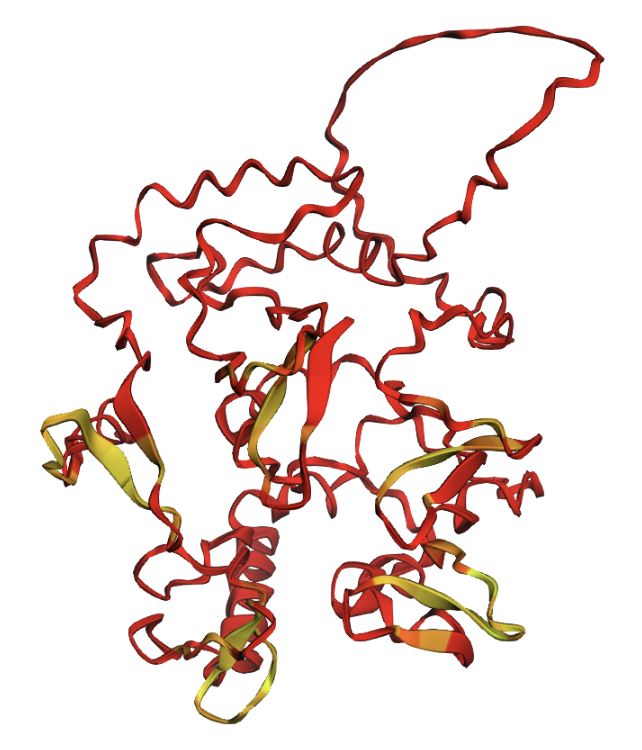 Positions 200–230 replaced with all glycines. A large disordered loop appears where the glycines were inserted.
Positions 200–230 replaced with all glycines. A large disordered loop appears where the glycines were inserted.
Single mutations had essentially no effect on the predicted structure where pTM remained identical at 0.157 and pLDDT changed by less than 0.5 points. Even replacing 31 consecutive residues with alanines or glycines only reduced pLDDT by 0.5–2.7 points. The glycine segment had the largest effect (pLDDT dropped from 39.15 to 36.48), likely because glycine is the most flexible residue and further destabilized an already disordered region. The structure appears highly resilient to mutation because there is so little predicted structure to begin with. ESMFold already has near-zero confidence in the original sequence, so mutations cannot meaningfully change much.
C3. Protein Generation (Inverse Folding)
I used ProteinMPNN (v_48_020, sampling temperature 0.1) to perform inverse folding on the AlphaFold-predicted backbone of Reflectin A1.
 ProteinMPNN amino acid probabilities at each position. Bright spots indicate positions where the model is highly confident about which residue should occupy that position.
ProteinMPNN amino acid probabilities at each position. Bright spots indicate positions where the model is highly confident about which residue should occupy that position.
The designed sequence had a sequence recovery of only 16.9%, meaning ProteinMPNN’s ideal sequence for this backbone is extremely different to the actual reflectin sequence. Reflectin sequence is optimized for self-assembly and dynamic nanostructure formation by sticking to other reflectin molecules. Many reflectin proteins come together and stack into organized layers, and it’s those layers that create the structural color (iridescence) in squid skin. The spacing between layers determines what wavelength of light gets reflected. ProteinMPNN designs for structural stability rather than the intermolecular interactions.
Folding the Designed Sequence with ESMFold
I folded the ProteinMPNN-designed sequence with ESMFold to compare it to the original:
| pTM | pLDDT | |
|---|---|---|
| Original Reflectin A1 | 0.157 | 39.15 |
| ProteinMPNN-designed sequence | 0.173 | 72.86 |
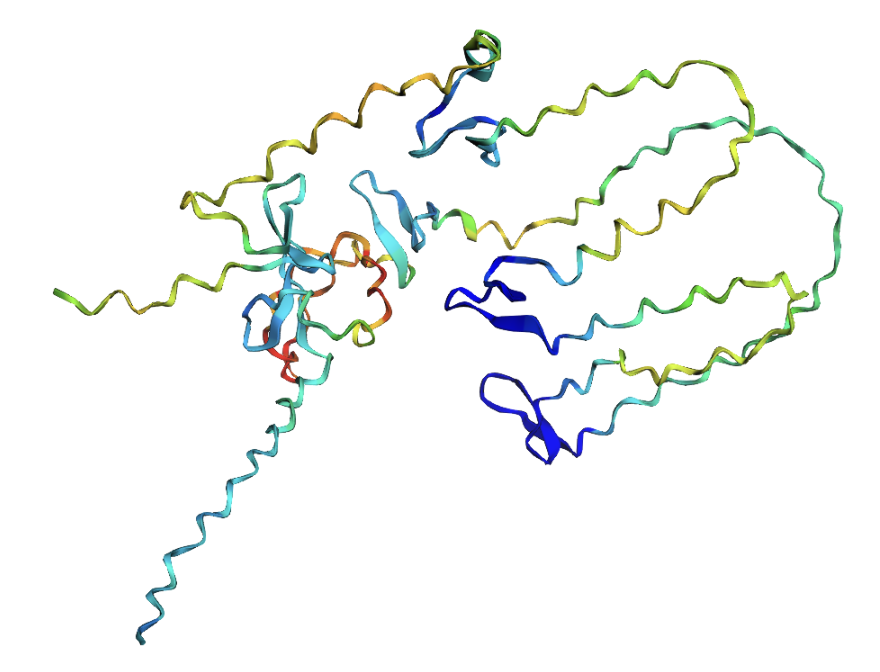 ESMFold prediction of the ProteinMPNN-designed sequence. The structure shows substantially more secondary structure elements (helices and sheets) compared to the original.
ESMFold prediction of the ProteinMPNN-designed sequence. The structure shows substantially more secondary structure elements (helices and sheets) compared to the original.
The ProteinMPNN-designed sequence folds with dramatically higher confidence (pLDDT 72.86 vs 39.15). The designed sequence contains many more prolines, glycines, and lysines residues that promote stable secondary structures and far fewer of reflectin’s characteristic methionine-rich and aromatic motifs. This highlights a fundamental tension in protein engineering: a sequence that folds well as a monomer is not necessarily a sequence that functions correctly.