Week 5: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Design of short peptides targeting the A4V mutant of human SOD1 (P00441) as a potential therapeutic strategy for familial ALS.
Part 1: Generate Binders with PepMLM
SOD1 Original Sequence:
UniProt ID: P00441
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQA4V Mutant SOD1 Sequence
UniProt ID: P00441 | Mutation: A4V (Ala→Val at position 5 in full sequence)
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQBinder Results
Four 12-mer peptides were generated by PepMLM-650M, and the known SOD1-binding peptide FLYRWLPSRRGG was scored separately for comparison.

| # | Peptide Sequence | Pseudo-Perplexity ↓ | Source | Notes |
|---|---|---|---|---|
| 1 | WHYYAVVARHKE | 21.55 | PepMLM generated | Highest perplexity of generated set |
| 2 | WRVGAVGVAHKK | 10.55 | PepMLM generated | Best confidence |
| 3 | WHSYATVVEHWE | 17.12 | PepMLM generated | — |
| 4 | WHYYVAGLEHKX | 14.75 | PepMLM generated | ⚠️ Contains ambiguous X residue |
| 5 | FLYRWLPSRRGG | 20.64 | Known binder | Reference benchmark |
Part 2: Evaluate Binders with AlphaFold3
Each peptide was co-folded with the A4V mutant SOD1 sequence as separate chains using the AlphaFold3 server. Peptide 4 (WHYYVAGLEHKX) could not be submitted due to the ambiguous X residue.
Peptide 1: WHYYAVVARHKE (ipTM = 0.32)
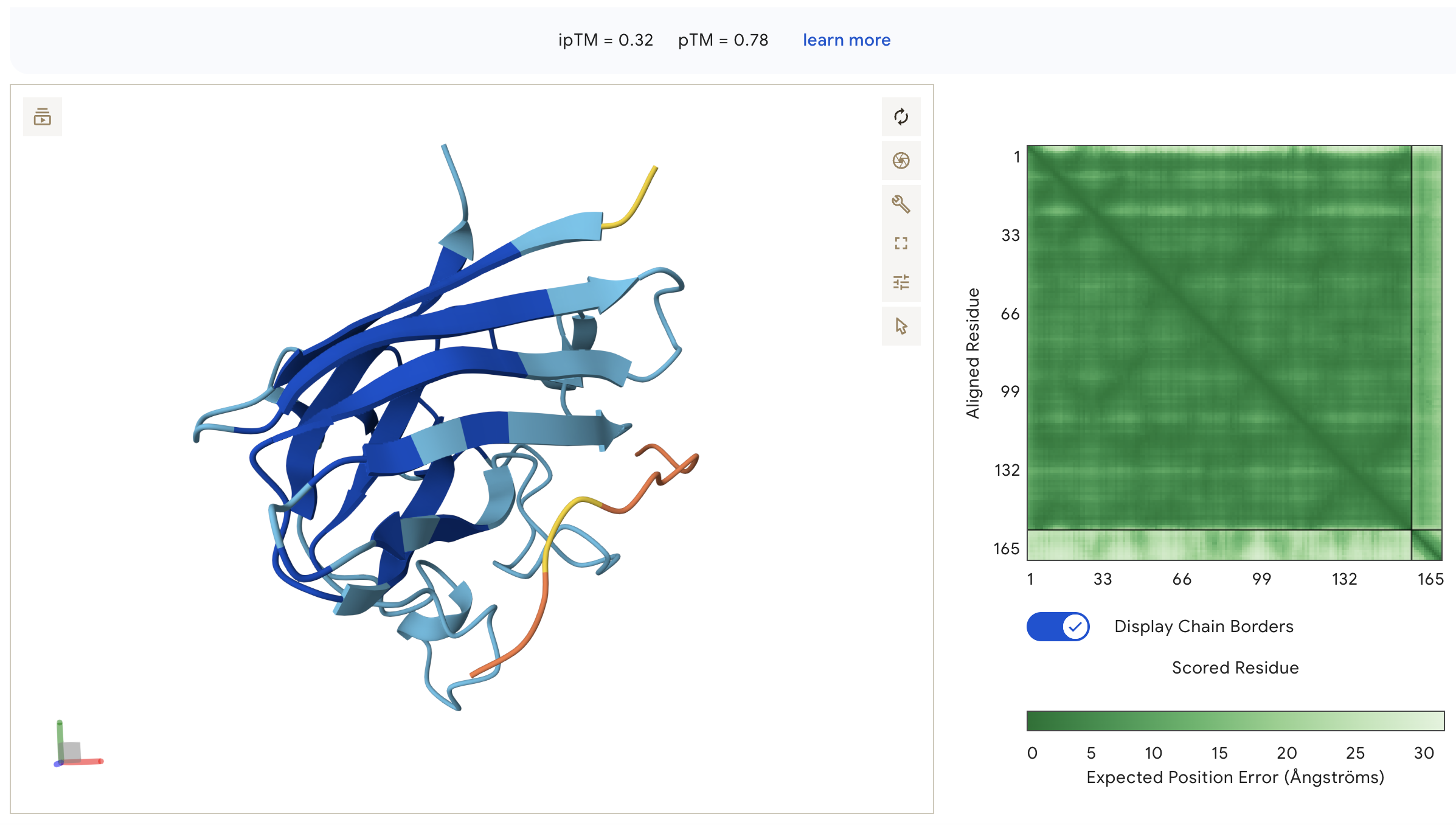
WHYYAVVARHKE: ipTM = 0.32, peptide appears as an unstructured surface-bound loop that is not contacting the N-terminus where A4V sits, with high inter-chain PAE indicating poorly defined binding.
Peptide 2: WRVGAVGVAHKK (ipTM = 0.49)
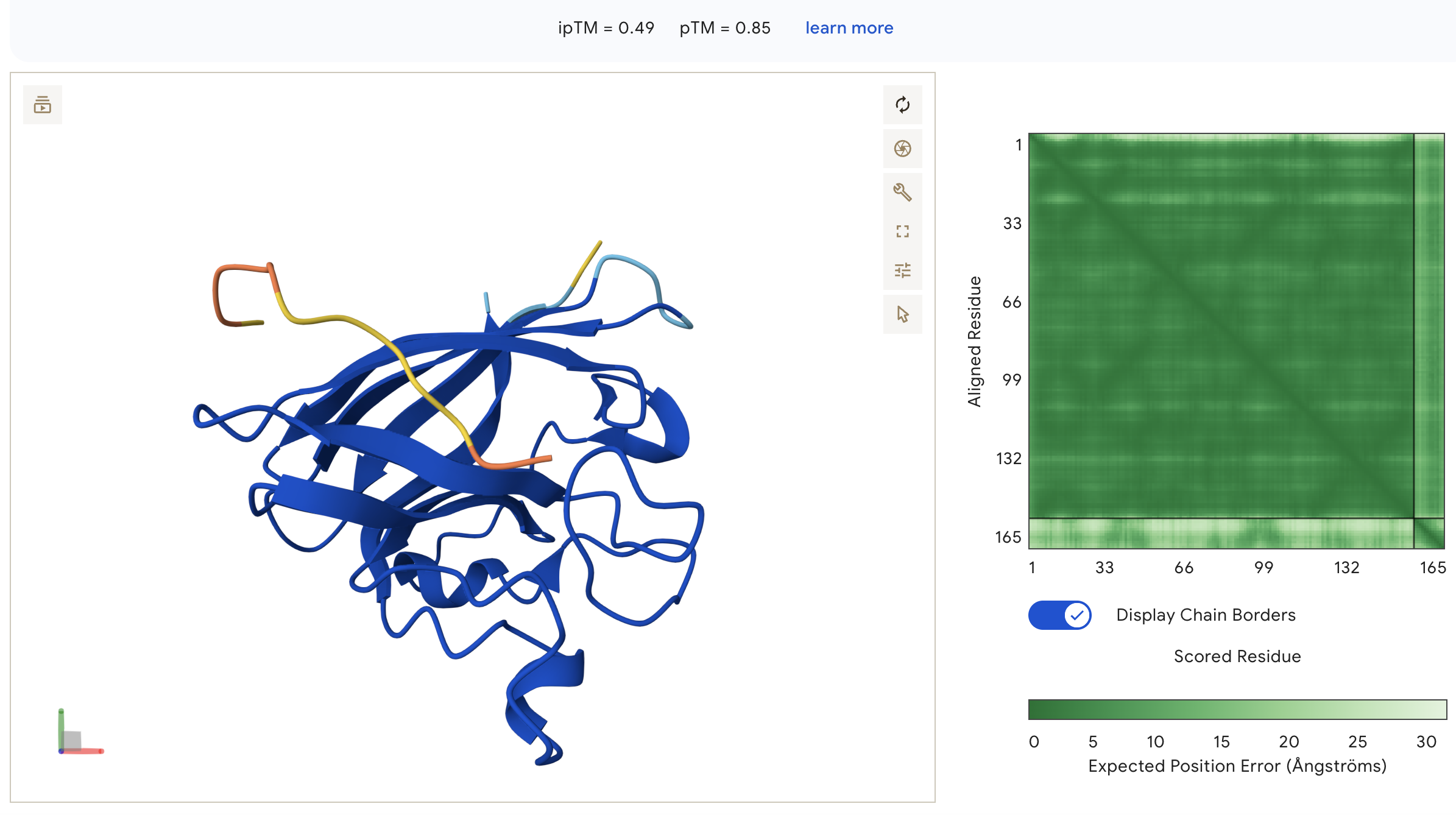
WRVGAVGVAHKK: ipTM = 0.49, peptide appears as an unstructured loop on the opposite face from the N-terminus, with high inter-chain PAE indicating poorly defined binding, though ipTM is notably higher than all other peptides including the known binder.
Peptide 3: WHSYATVVEHWE (ipTM = 0.28)
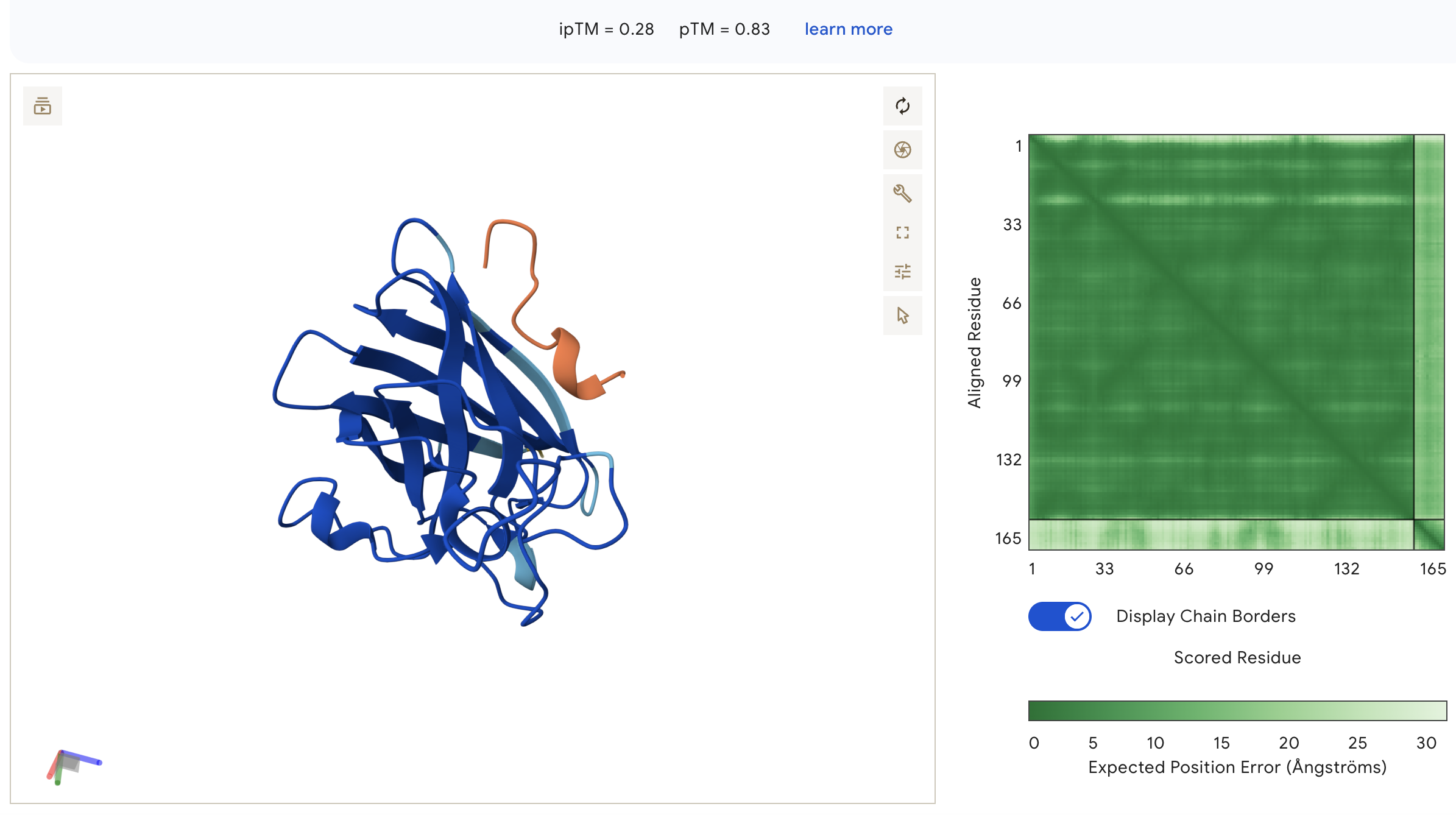
WHSYATVVEHWE: ipTM = 0.28, peptide is surface-bound on the lateral face of the β-barrel away from the N-terminus, with very light inter-chain PAE indicating the weakest binding confidence of all peptides.
Known Binder: FLYRWLPSRRGG (ipTM = 0.33)
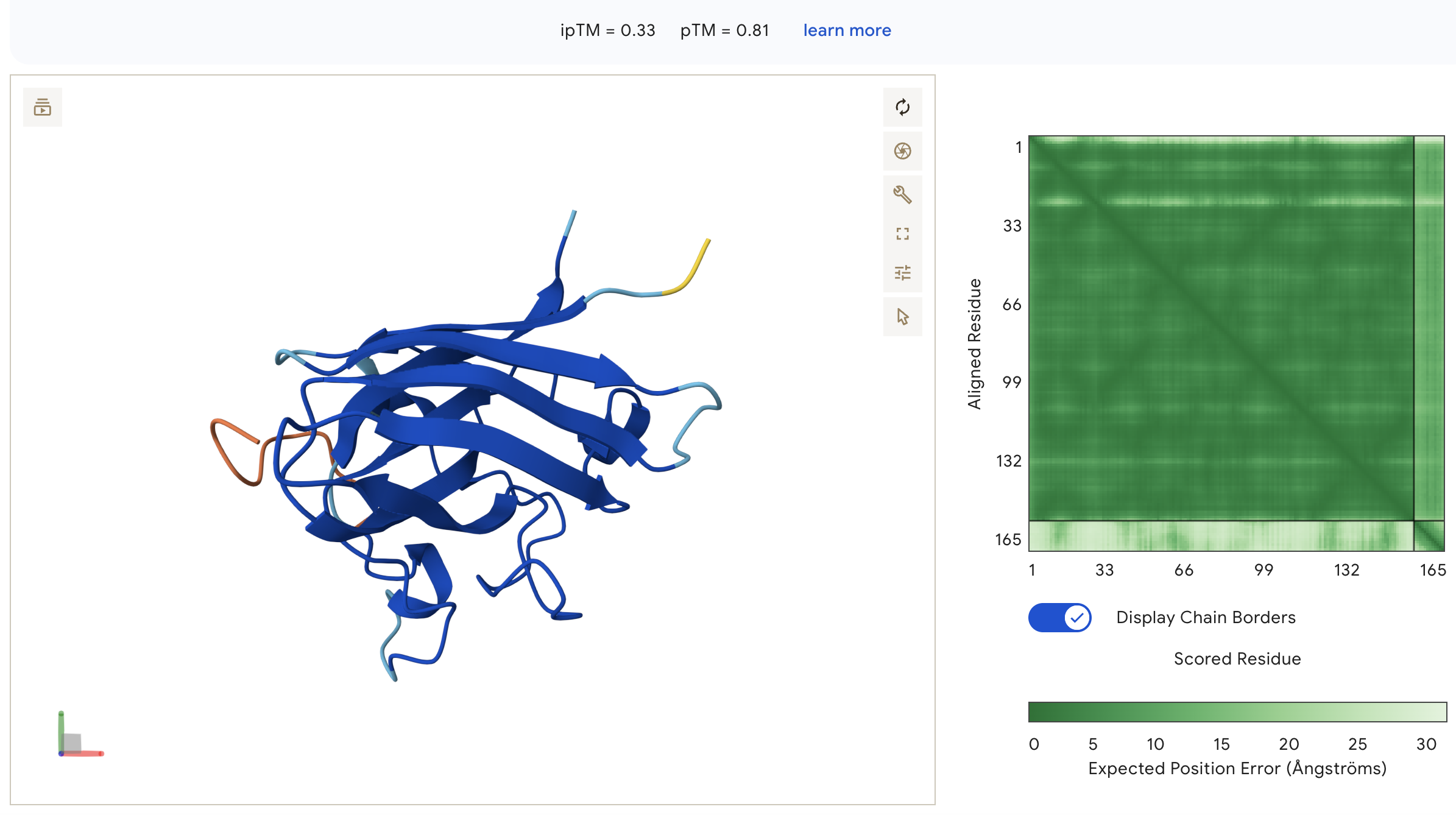
FLYRWLPSRRGG: ipTM = 0.33, peptide wraps around the lateral face of the β-barrel as an extended unstructured loop, with no engagement near the N-terminus and high inter-chain PAE indicating poorly defined binding geometry.
| # | Peptide | ipTM | pTM | Notes |
|---|---|---|---|---|
| 1 | WHYYAVVARHKE | 0.32 | 0.78 | Surface-bound, distal from N-terminus |
| 2 | WRVGAVGVAHKK | 0.49 | 0.85 | Best ipTM, exceeds known binder |
| 3 | WHSYATVVEHWE | 0.28 | 0.83 | Lowest ipTM, lateral surface binding |
| 4 | WHYYVAGLEHKX | — | — | Skipped — invalid X residue |
| 5 | FLYRWLPSRRGG | 0.33 | 0.81 | Known binder reference |
All peptides scored below 0.5 ipTM, indicating low overall interface confidence across the set; however, peptide 2 (WRVGAVGVAHKK) achieved the highest ipTM at 0.49, exceeding the known binder (0.33), suggesting it is the strongest structural candidate despite none localizing clearly to the N-terminal A4V site.
Part 3: Evaluate Properties with PeptiVerse
Each peptide was evaluated against the A4V mutant SOD1 sequence using PeptiVerse for predicted binding affinity, solubility, hemolysis probability, net charge, and molecular weight.
Peptide 1: WHYYAVVARHKE

Peptide 2: WRVGAVGVAHKK

Peptide 3: WHSYATVVEHWE

Known Binder: FLYRWLPSRRGG

| # | Peptide | Binding Affinity (pKd/pKi) | Solubility | Hemolysis | MW (Da) | Net Charge (pH 7) |
|---|---|---|---|---|---|---|
| 1 | WHYYAVVARHKE | 5.505 | Soluble | Non-hemolytic (0.040) | 1558.7 | +0.94 |
| 2 | WRVGAVGVAHKK | 5.520 | Soluble | Non-hemolytic (0.020) | 1307.5 | +2.85 |
| 3 | WHSYATVVEHWE | 5.604 | Soluble | Non-hemolytic (0.056) | 1543.6 | −2.06 |
| 4 | WHYYVAGLEHKX | — | — | — | — | — |
| 5 | FLYRWLPSRRGG | 5.968 | Soluble | Non-hemolytic (0.047) | 1507.7 | +2.76 |
Higher ipTM does not correlate with stronger predicted binding affinity; peptide 3 (WHSYATVVEHWE) shows the highest affinity (5.604) despite the lowest ipTM (0.28), while peptide 2 (WRVGAVGVAHKK) has the best structural confidence but mid-range affinity (5.520); all peptides are predicted soluble and non-hemolytic, making peptide 2 the best overall candidate as it balances the strongest structural binding (ipTM 0.49), low hemolysis risk (0.020), and relatively good binding affinity.
Part 4: Motif-Guided Generation with moPPIt
moPPIt was run targeting motif positions 1–8 of A4V SOD1, with objectives set to Hemolysis, Solubility, Affinity, and Motif (all weights = 1). Four 12-mer peptides were generated.
| # | Peptide | Hemolysis ↑ | Solubility ↑ | Affinity ↑ | Motif Score ↑ |
|---|---|---|---|---|---|
| 1 | HTPYSPYTCKNI | 0.919 | 0.750 | 6.28 | 0.822 |
| 2 | DTDDTKPGWTCW | 0.959 | 0.750 | 6.69 | 0.754 |
| 3 | EKASGGHEHNPI | 0.940 | 0.750 | 5.08 | 0.364 |
| 4 | KKFQEVYRKKTC | 0.955 | 0.833 | 6.90 | 0.706 |
Comparison and Evaluation
PepMLM peptides are dominated by aromatic residues (W, H, Y, F), while moPPIt peptides are more compositionally diverse featuring charged and polar residues alongside three cysteine-containing sequences because generation was guided by multiple objectives. The moPPIt set achieves higher predicted affinity overall, though motif adherence varies: EKASGGHEHNPI scores only 0.364. The computational scores are predictions, not measurements so the first step is to actually test whether these peptides bind A4V SOD1 in the lab and confirm the predicted properties hold experimentally. From there, you would test whether binding has a functional effect in cells and what type of delivery method.
Part C: L-Protein Mutant Design
The MS2 lysis protein (UniProt P03609) is a 75-residue protein with a soluble N-terminal domain (residues 1–40) that interacts with the E. coli chaperone DnaJ, and a transmembrane domain (residues 41–75) responsible for membrane poration and lysis. The goal is to engineer mutants that improve DnaJ-independence or lysis efficiency to overcome bacterial resistance.
Mutation Selection
Five mutants were selected using three sources: (1) experimental lysis data from Chamakura et al., (2) ESM-2 log-likelihood ratio (LLR) scores from the Colab notebook, and (3) conservation analysis across 26 homologous sequences from pBLAST.
Mutations were selected where experimental data confirmed both Lysis=1 and Protein Levels=1, meaning the mutant phage still kills bacteria and still produces the protein. Conserved positions with strong experimental support were also included.
| # | Mutation | Region | Variable in Nature? | Exp. Lysis | Exp. Protein | ESM-2 LLR |
|---|---|---|---|---|---|---|
| 1 | S15A | Soluble | Yes | 1 | 1 | +0.04 |
| 2 | R18G | Soluble | Conserved | 1 | 1 | −0.85 |
| 3 | R30Q | Soluble | Conserved | 1 | 1 | −0.37 |
| 4 | L44P | Transmembrane | Conserved | 1 | 1 | −1.59 |
| 5 | A45P | Transmembrane | Yes | 1 | 1 | +0.04 |
The ESM-2 LLR scores weakly correlate with the experimental lysis data. S15A and A45P have near-zero LLR scores (tolerated but not predicted as beneficial), while R18G, R30Q, and L44P are all negative where the model predicts these as slightly harmful. Yet all five maintain lysis experimentally.
S15A: position 15 is variable across MS2 strains, and A is observed in nature at this position, making this the most conservative and well-supported mutation. Located in the soluble domain, it may alter DnaJ interaction.
R18G and R30Q: both in the soluble domain at conserved positions, but directly confirmed by experimental data to maintain lysis function.
L44P and A45P: in the transmembrane domain. Both are experimentally confirmed to maintain lysis (Lysis=1, Protein=1) with only A45P variable in nature.