Week 10: LC-MS at Waters Immerse Cambridge
Overview
We rotated through three stations at Waters Immerse Cambridge to characterize proteins by mass spectrometry. The first two stations used eGFP (a 27 kDa fluorescent protein); the third used KLH, a much larger protein complex.
- Xevo G3 QTof: intact mass and native vs. denatured structure
- BioAccord: peptide mapping for amino acid sequence
- Xevo CDMS: megadalton-sized protein complexes
Sample Prep
Buffer-exchanged eGFP into 50 mM ammonium acetate using BioRad Micro Bio-Spin columns. Ammonium acetate is the standard MS-compatible buffer because it evaporates cleanly in the electrospray source.
Pipetted ammonium acetate into the wash tubes.
Snapped the bottom off each spin column, drained the packing buffer in a mini centrifuge, and washed four times with ammonium acetate.

Loaded 50 µL of eGFP onto the column and centrifuged for 4 minutes at 1000 x RCF on the Eppendorf 5424 R to elute the buffer-exchanged protein. Passed the eluate through a second column for cleanup.

Diluted 10x, transferred to autosampler vials, split into aliquots for the three stations.

Station 1: Xevo G3 QTof — Intact Mass and Structure
Intact mass (LC-MS). Loaded the autosampler vials into the Waters Acquity Premier UPLC. The C4 column with acidic mobile phases unfolded the protein and pushed it through the mass spec as an extended chain, producing a wide ladder of charge states. Back-calculating from the peak spacing confirmed eGFP at ~27 kDa.


Native vs. denatured (direct infusion). No LC for this part. Loaded the same sample into a glass syringe and infused directly at 10 µL/min.
- Native (ammonium acetate, neutral pH): folded eGFP, low charge states in a narrow distribution.
- Denatured (added 5 µL formic acid): unfolded eGFP, high charge states in a much broader distribution.
The takeaway: charge state distribution reports on folding, not just mass.
Station 2: BioAccord — Peptide Mapping
Bottom-up sequencing of eGFP. Workflow:
- Denature in 7 M guanidine HCl
- Reduce with 5 mM DTT at 55°C for 5 minutes
- Buffer exchange into Tris-HCl/CaCl2 with another Bio-Spin column
- Digest with trypsin at 55°C for 20 minutes (cleaves after every K or R)
- Quench with 1% formic acid
- Inject into the BioAccord LC-MS
The BioAccord separated peptides by hydrophobicity on a C18 column, then for each peak measured the peptide’s mass plus its fragmentation pattern in the collision cell. Stitching identified peptides back together reconstructed ~88% of the eGFP sequence.
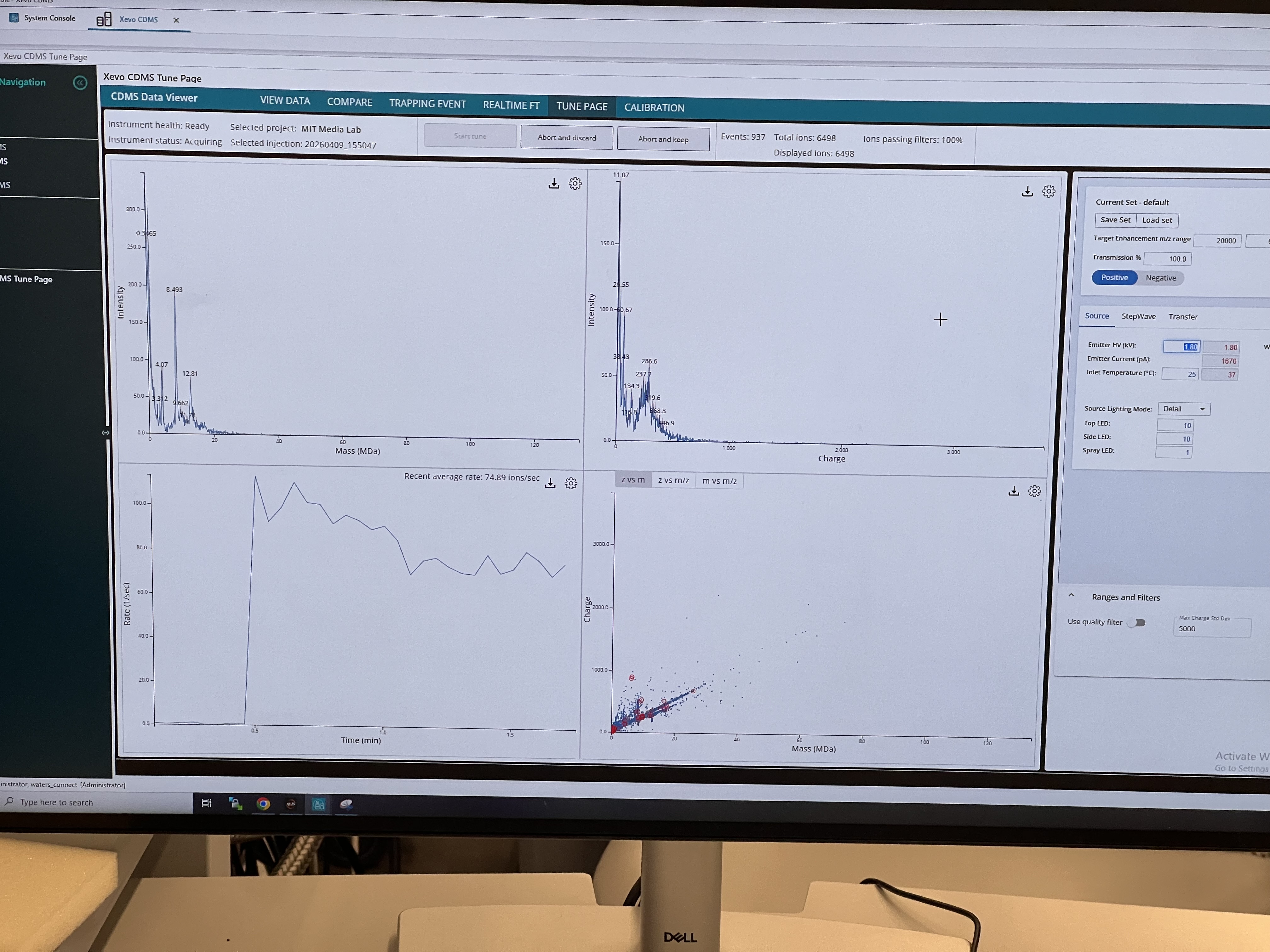
Station 3: Xevo CDMS — Megadalton Complexes
CDMS measures the charge of each individual ion alongside its m/z, so you can solve for mass one ion at a time. Useful for very large protein complexes where conventional MS gives unresolved peaks because the charge states are too close together.
Worked with KLH (a marine mollusk protein) directly infused via glass syringe.


Built up the mass distribution by accumulating ions in the Tune Page until we reached ~1500 displayed ions. Saw distinct peaks at ~4 MDa (decamer) and 8–13 MDa (stacked multi-decamers).