Week 4 HW: Protein Design Part I
⛓️ Collab Notebook Link
⛓️ Link to the Colab for the chosen protein (FOXO3): Collab notebook
💡 PART A - CONCEPTUAL QUESTIONS
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Avogadro’s number: 1 mole of anything = 6.022 * 10^23 molecules. Since 1 Da = 1 g/mol, a molecule with mass 100 Da has a molar mass of 100 g/mol.
so: 500g / (100g/mol) = 5 mo * 6.022*(10^23) = 3.01e+24 molecules
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The cells and proteins contained in the food are completely broken down into their most essential forms (ie amino acids) before being absorbed and used by the body. The proteins contained in the food are not used directly, instead they are broken down into amino acids which are then used as building materials when our cellular machine reads our own DNA to build the proteins we need (which might be different than the versions encoded by other species).
Why are there only 20 natural amino acids?
A “why” question can never be rigurously answered in biology, but we can infer that this set of amino acids contains all the necessary properties to create life.
(Note: by doing this exercise, I learned that there are actually 22 amino acids in nature, with the 2 additional ones being selenocystein and pyrrolysine)
Can you make other non-natural amino acids? Design some new amino acids.Where did amino acids come from before enzymes that make them, and before life started?
In 1953, scientists Stanley Miller and Harold Urey were able to produce amino acids by simulating early Earth conditions, by using electric sparks in a mixture of water, ammonia, methane and hydrogen. Other possible sources are meteorites (which have been found to be rich in amino acids), hydrothermal vents, photochemistry (UV radiation on ice surfaces) and, potentially, Strecker synthesis.
If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed. They are the mirror image of L-amino acids, which are right-handed for the following reason: ”Natural L-amino acids have their φ (phi) and ψ (psi) backbone dihedral angles in a region of the Ramachandran plot (approximately φ = −60°, ψ = −40°) that produces a right-handed helix”
((Curious fact: origin of the “D” and “L” I just learned)) (nested/click to see)
“The letters L and D come from an old naming system that compares molecules to the simple sugar glyceraldehyde and to “left/right” in Latin.
Historical origin: Chemists first defined D‑ and L‑ using glyceraldehyde: in a standard Fischer projection, the isomer with the key group on the right was called D (from dextro = right), and the one with it on the left was called L (from laevo = left).
Can you discover additional helices in proteins?
Yes, I learned that there’s other types like n-helix, collagen triple helix,
Why are most molecular helices right-handed?
Why do β-sheets tend to aggregate?
Because the edge strands of every β-sheet have unsatisfied hydrogen bond capacity, and are therefore “looking” for more strands to bond with. - What is the driving force for β-sheet aggregation?
The primary driver is hydrogen bonding. Each hydrogen bond contributes ~2-8kJ/mol, so a sheet with many bonds has a large energetic benefit.
Why do many amyloid diseases form β-sheets?
”Amyloids are ordered protein aggregates with a characteristic cross-β structure: β-strands run perpendicular to the fibril long axis, forming a “spine” of stacked sheets. They are extraordinarily stable: resistant to heat, most detergents, and proteases.”
- Can you use amyloid β-sheets as materials?
-
Yes. Some examples I have found: spider silk, Curli fibers, biotemplating for metallic wires.
13. Design a β-sheet motif that forms a well-ordered structure.
💡 Part B: Protein Analysis and Visualization
1 Briefly describe the protein you selected and why you selected it.
FOXO3 for its suspected longevity benefits. Certain alleles are found at higher frequencies in supercentenarians.
2 Identify the amino acid sequence of your protein.
• How long is it? What is the most frequent amino acid?
673 amino acids (states in the UniProt page)
• How many protein sequence homologs are there for your protein?
250 homologs
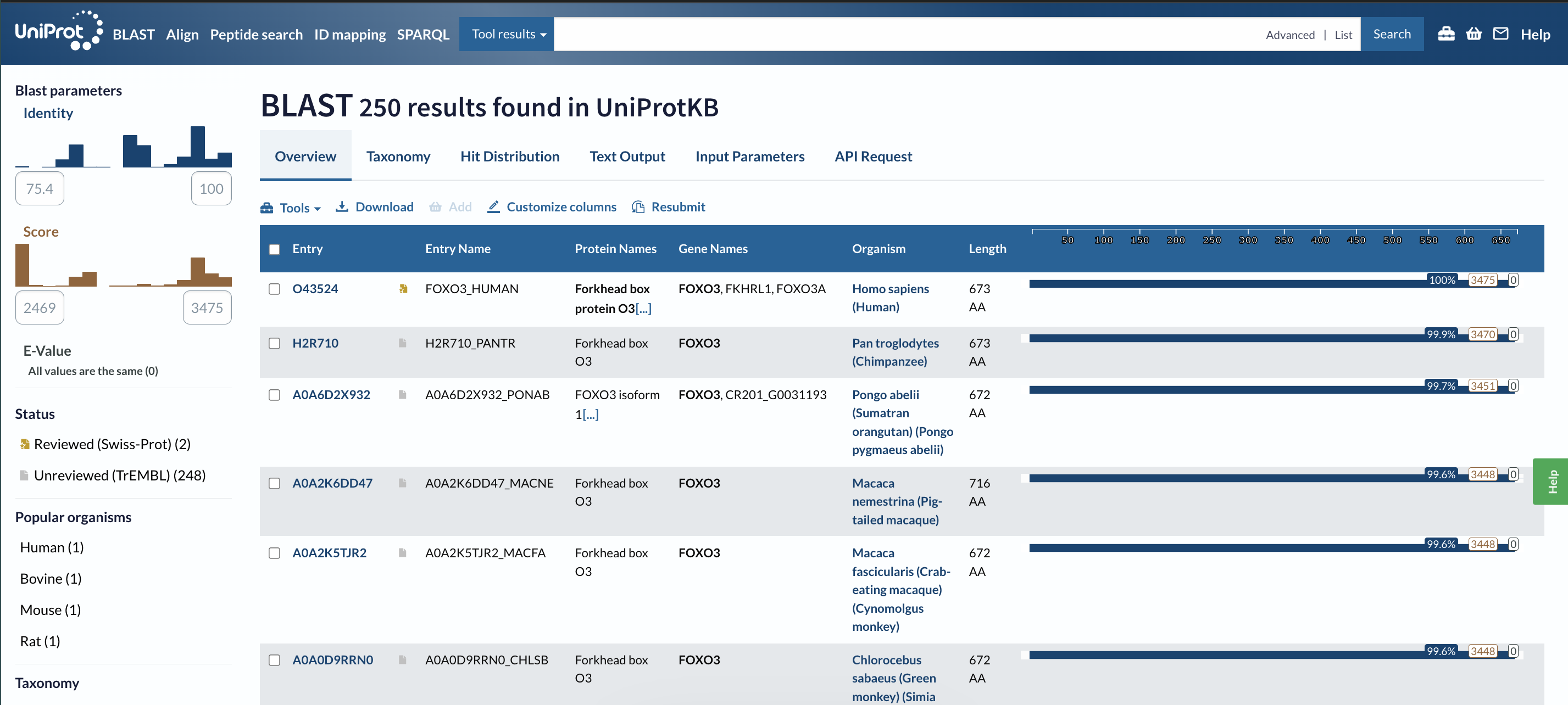
• Does your protein belong to any protein family?
”member of the forkhead box class O (FOXO) subfamily” (from RCSB)
3 Identify the structure page of your protein in RCSB • When was the structure solved? Is it a good quality structure?
The entry was submitted in 2008.
(page: https://www.rcsb.org/structure/2K86) (I couldn’t find this on the page, so I asked AI for help and it said: “Experimental method is solution NMR, so there is no crystallographic resolution value like “2.7 Å”; resolution only applies to diffraction/EM, not to NMR”)
• Are there any other molecules in the solved structure apart from protein?
There’s no other molecules present (according to this page). • Does your protein belong to any structure classification family?
Under “family & domains” in UniProt it says “Winged helix-like DNA-binding domain superfamily/Winged helix DNA-binding domain 1 hit”
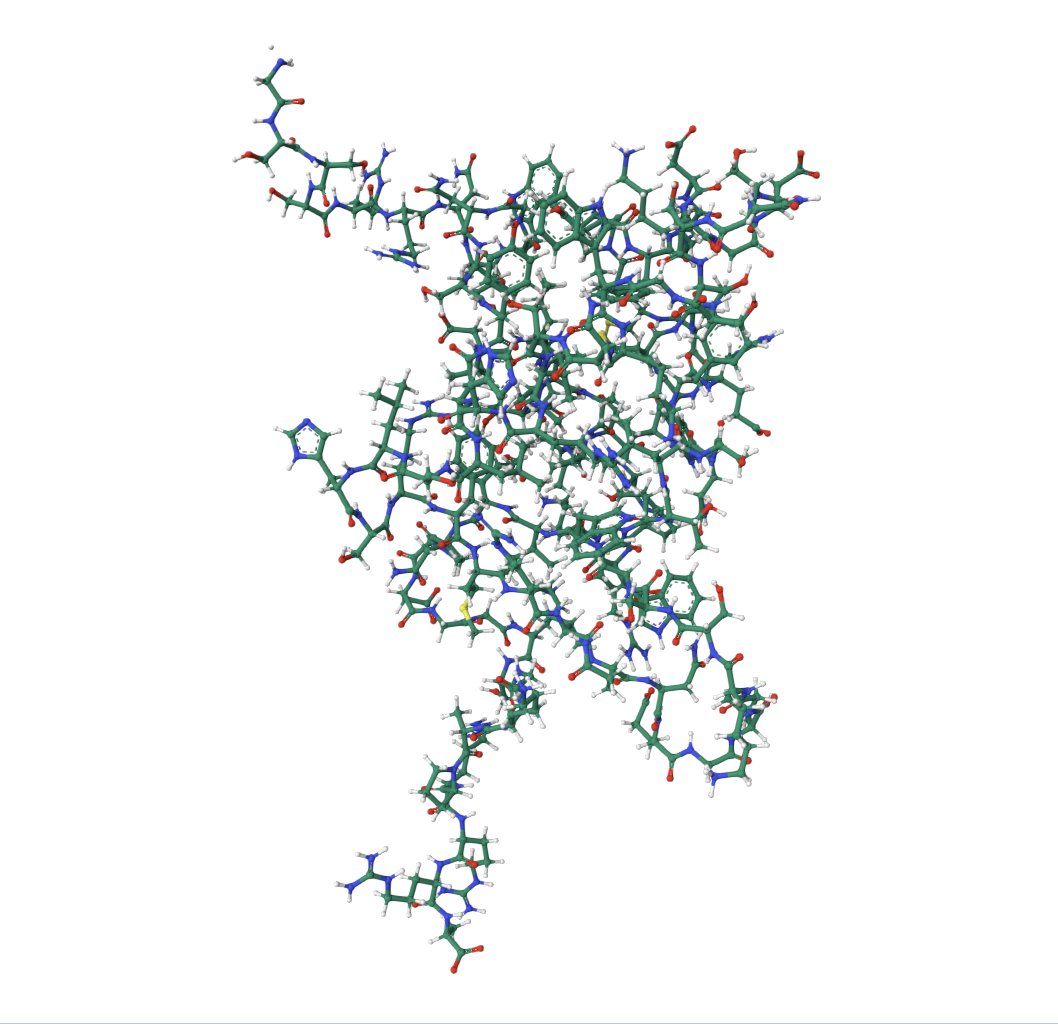
4 Open the structure of your protein in any 3D molecule visualization software:
• Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
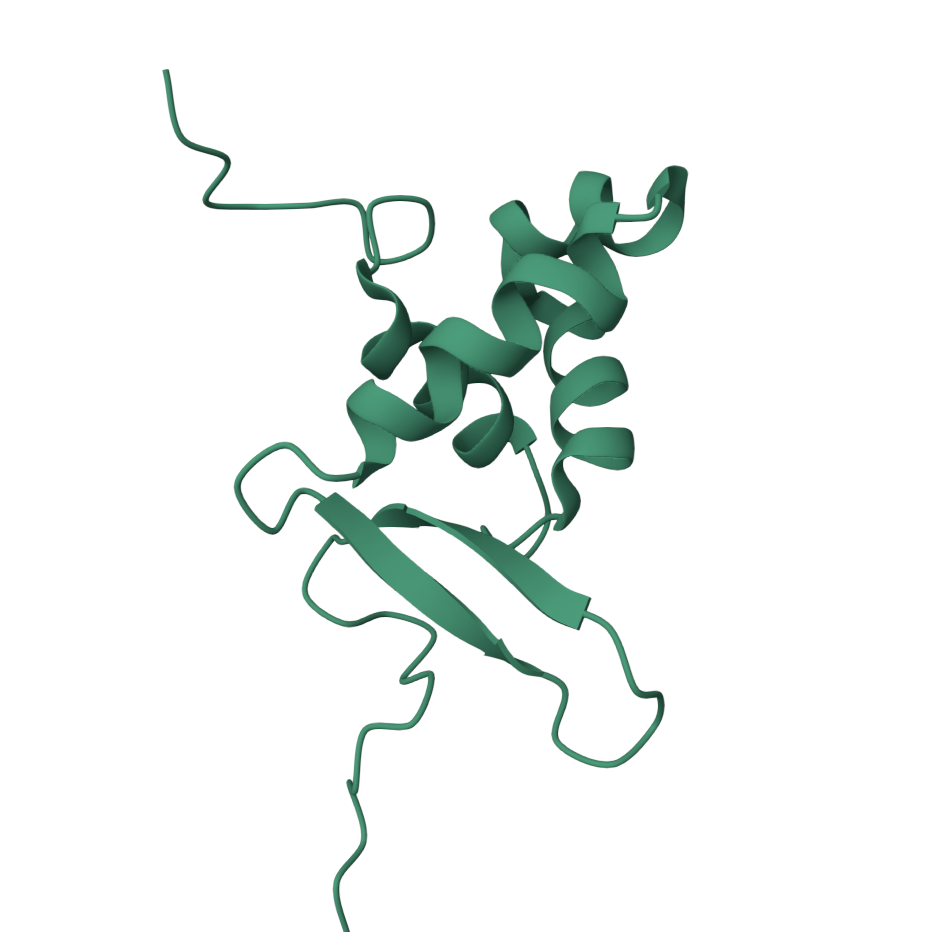

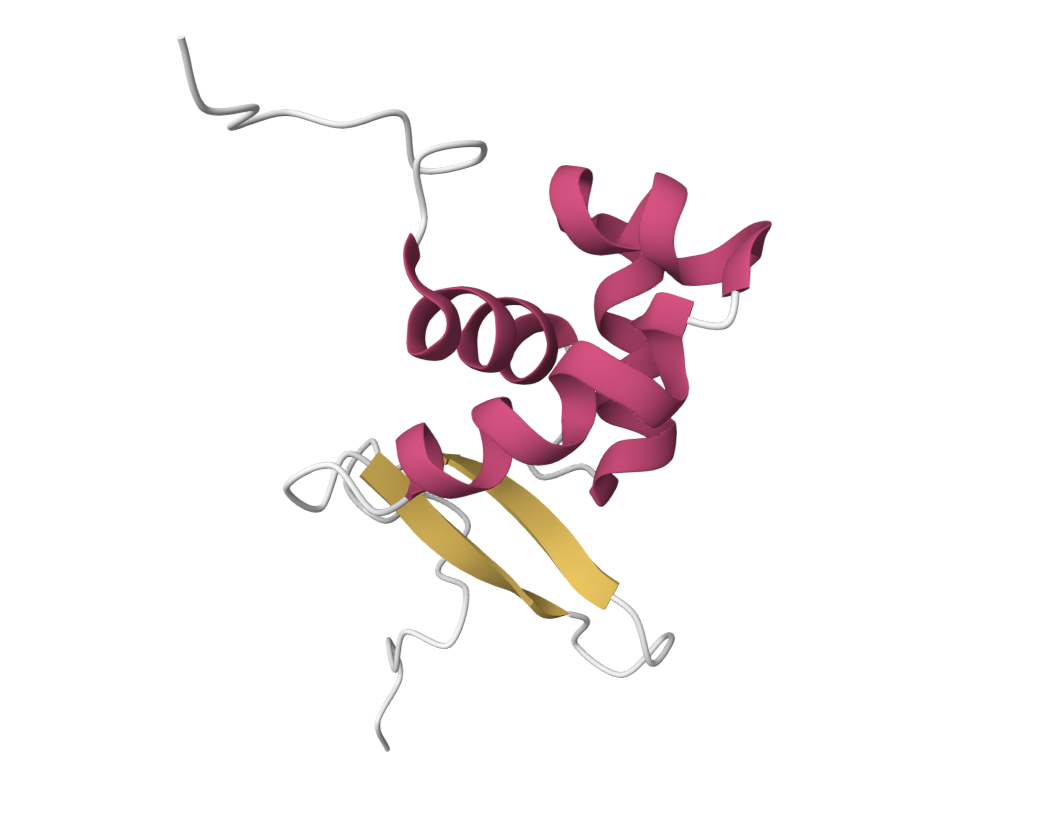
• Color the protein by secondary structure. Does it have more helices or sheets?
It has more helices.
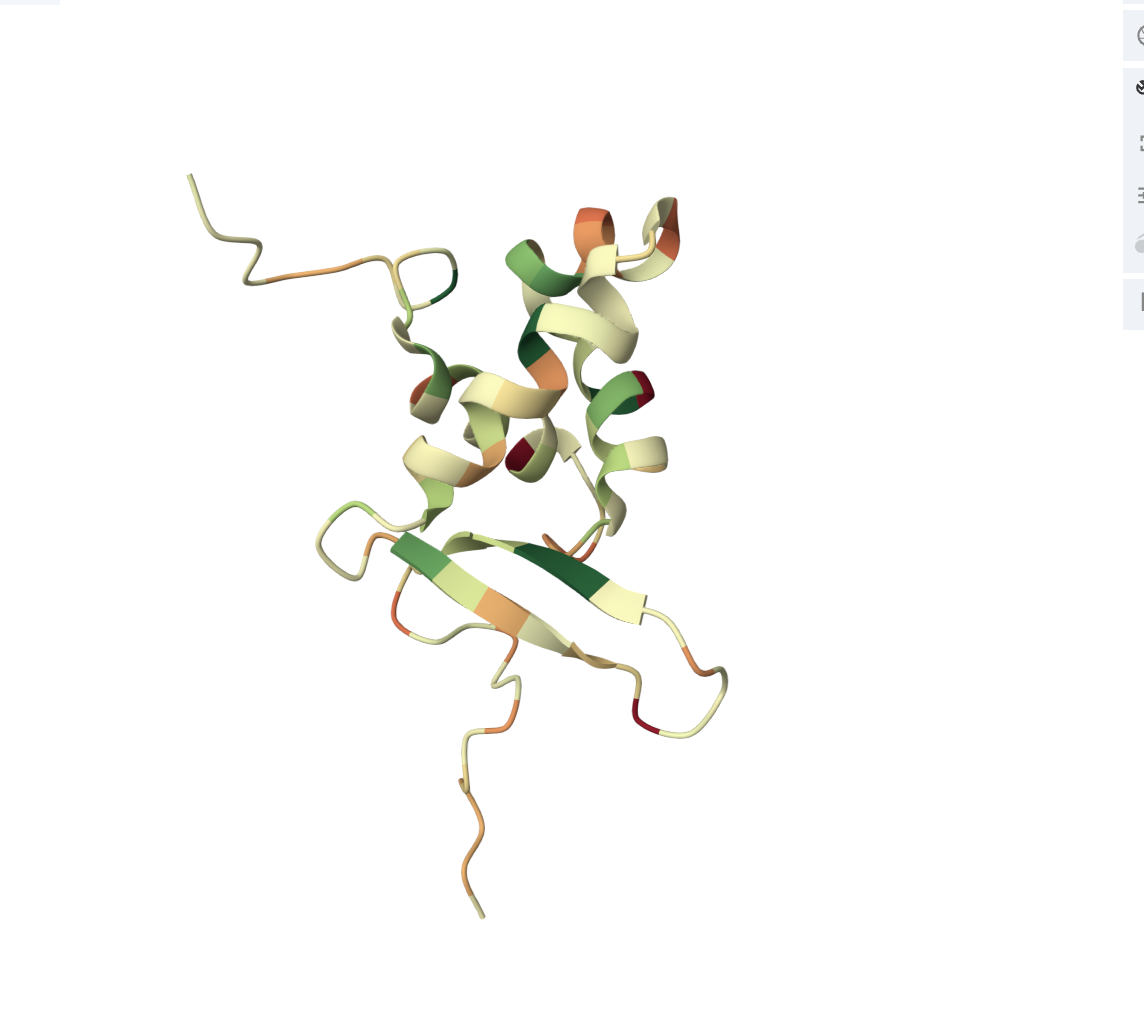
• Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The core of the protein (interior) is dominated by hydrophobic residues (green patches). This is the hydrophobic core, which drives protein folding (hydrophobic residues hide from water by burying inward). The outer loops and exposed regions show more orange/red, meaning hydrophilic/charged residues (these face outward toward the watery cellular environment).
• Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?

💡 C1. Protein Language Modeling
- Deep Mutational Scans
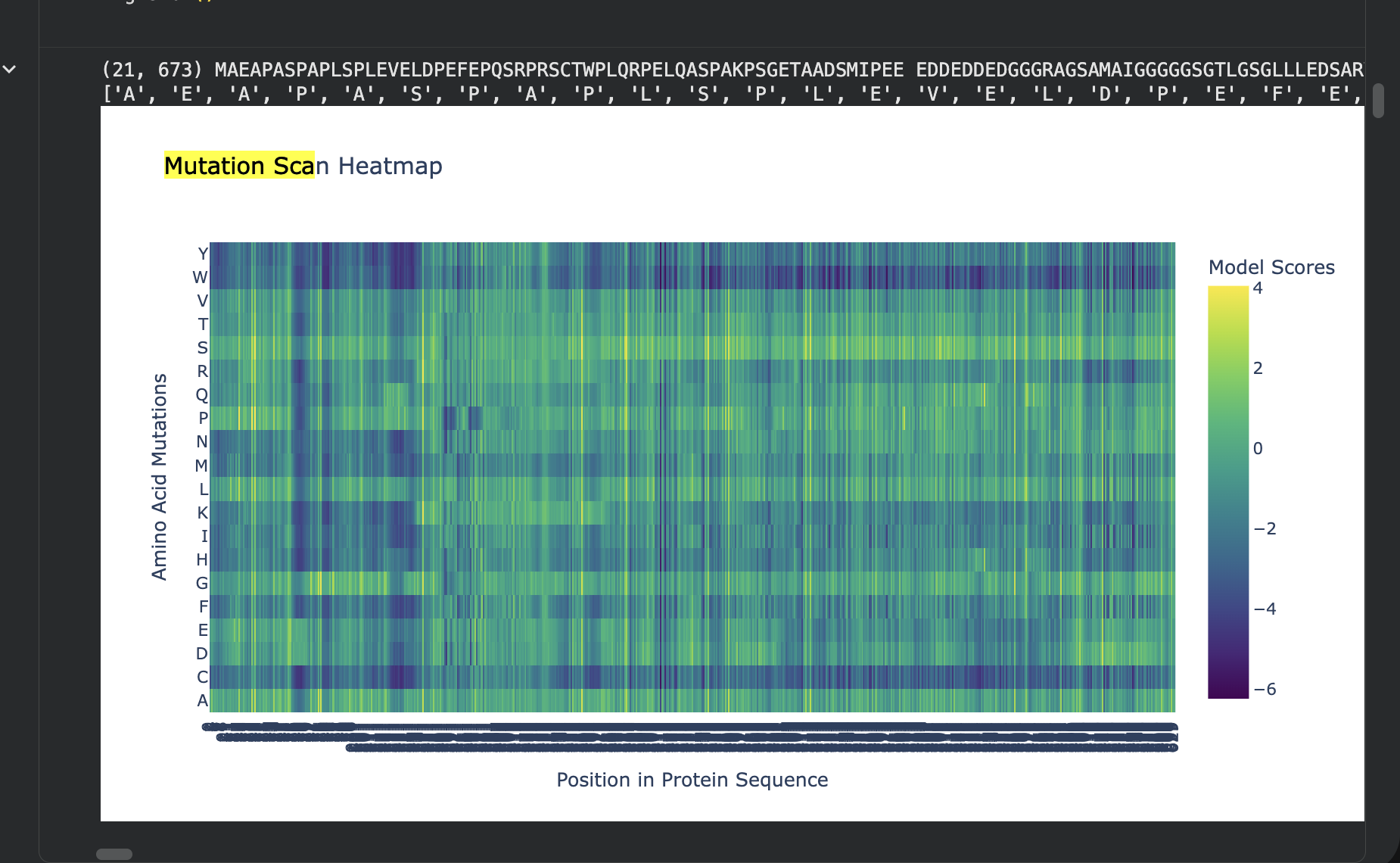
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The most disfavored mutation is:
Pos 1: M->W, LLR=-17.779
The most favored one:
Pos 261: T->S, LLR=3.991
Latent Space Analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
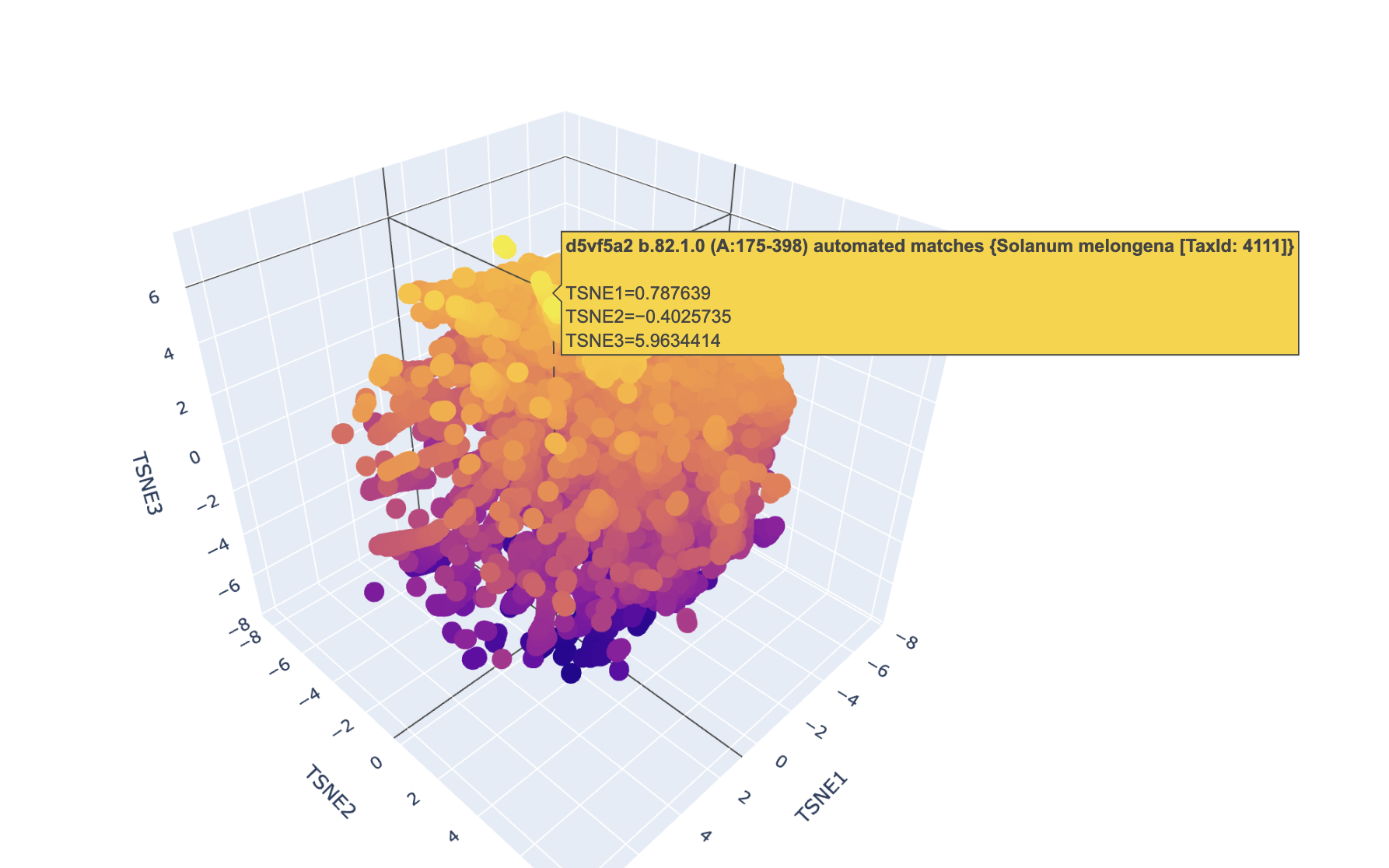
Analyze the different formed neighborhoods: do they approximate similar proteins?
I had trouble finding this protein due to its nature.
Place your protein in the resulting map and explain its position and similarity to its neighbors.