Week 6 HW: Genetic Circuits Part I
SECTION 1: PCR & GIBSON ASSEMBLY
“What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? It is composed of 3 essential components: 1. Phusion DNA Polymerase: a high-fidelity enzyme with low error rates (50x higher fidelity than Taq) and fast extension speeds (15-30sec/kilobase) 2. dNTPs (deoxynucleotide triphosates) (dATP, dCTP,dGTP, dTTP): the nucleotide building blocks the polymerase incorporates into the DNA starnd it builds 3. An optimized reaction buffer containing Mg(^2+) ions (polymerase cofactor that also stabilizes annealing) and KCl, which promotes specific primer binding while suppressing non-specific interactions. The Green Master Mix also contains other reagents (green dye, density reagents) so the reaction can be directly loaded into an agarose gel.
What are some factors that determine primer annealing temperature during PCR? It is determined by the melting temperature (Tm) of the primer, which is influenced by factors such as the primer length (longer = higher Tm), the GC content (more GC = higher Tm), and salt concentration in the reaction buffer.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. They are both methods for generating linear DNA fragments, with different strengths.
PCR is used to exponentially amplify a target region through cyclic multi-step iterations (denaturation -> annealing -> extension). It uses sequence-specific primers + a thermostable DNA polymerase to produce DNA fragments that match the primer binding sites, and can introduce mutations or add overlapping sequences (like the ones used in Gibson assembly). It is especially useful when we want to generate much larger quantities of a small sample of DNA.
Restriction enzyme digestion is preferred where fidelity is critical, i.e. when generating sticky ends for traditional ligation cloning or working on large constructs.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning? For this to be the case, the following conditions must be met:
- Homologous overlaps must exist between adjacent fragments (the 3’ end of one fragment must share 20-40 bp of sequence identity with the 5’ end of the next fragment)
- Template DNA must be removed
- The fragments must be purified (to remove enzymes, primers and nucleotides that could interfere with the Gibson reactions)
- The purified DNA concentration should be high enough to provide enough material for the Gibson reaction
How does the plasmid DNA enter the E. coli cells during transformation? For example, in the case of the heat shock method (used in the lab for this module), we pre-treat the cells with CaCl_2 (to neutralize the negative charges on the phospholipid membrane and the DNA backbone) to create “(chemically) competent cells”. We incubate both the cells and the plasmid DNA on ice for 30 minutes, then induce a 42’C heat shock for exactly 45 seconds to induce a thermal gradient which is believed to open transient pores in the cells’ membrane which allows the DNA to pass through, We finish by returning them to ice for 5 minutes so the membranes are sealed again, with the DNA now inside. Lastly, we incubate the cells in SOC-rich medium for 60 minutes (at 37ºC with shaking) to allow them to recover and keep dividing. Selection is applied by expressing the antibiotic resistance gene (only the cells that have taken up the DNA will survive in the new media with antibiotic).
ASIMOV KERNEL REPRESSILATOR
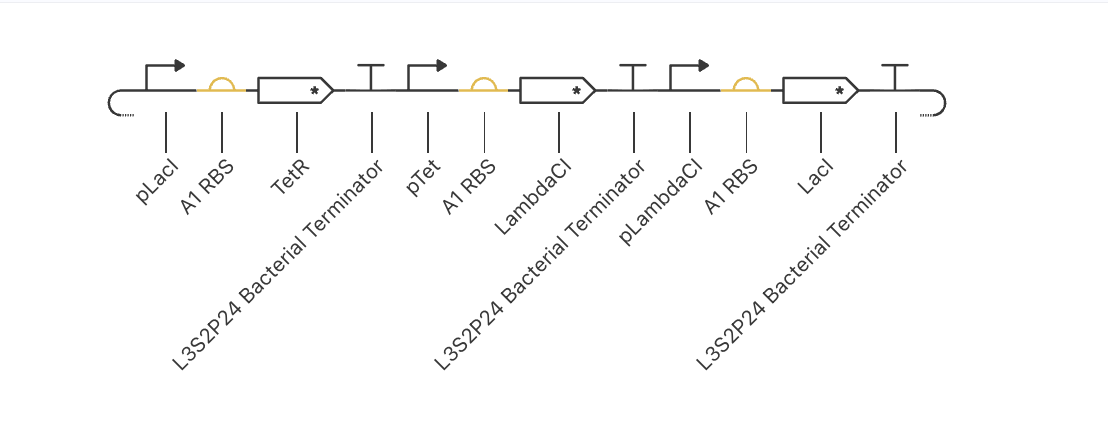
Original repressilator construct from the example repo, with its simulation data:
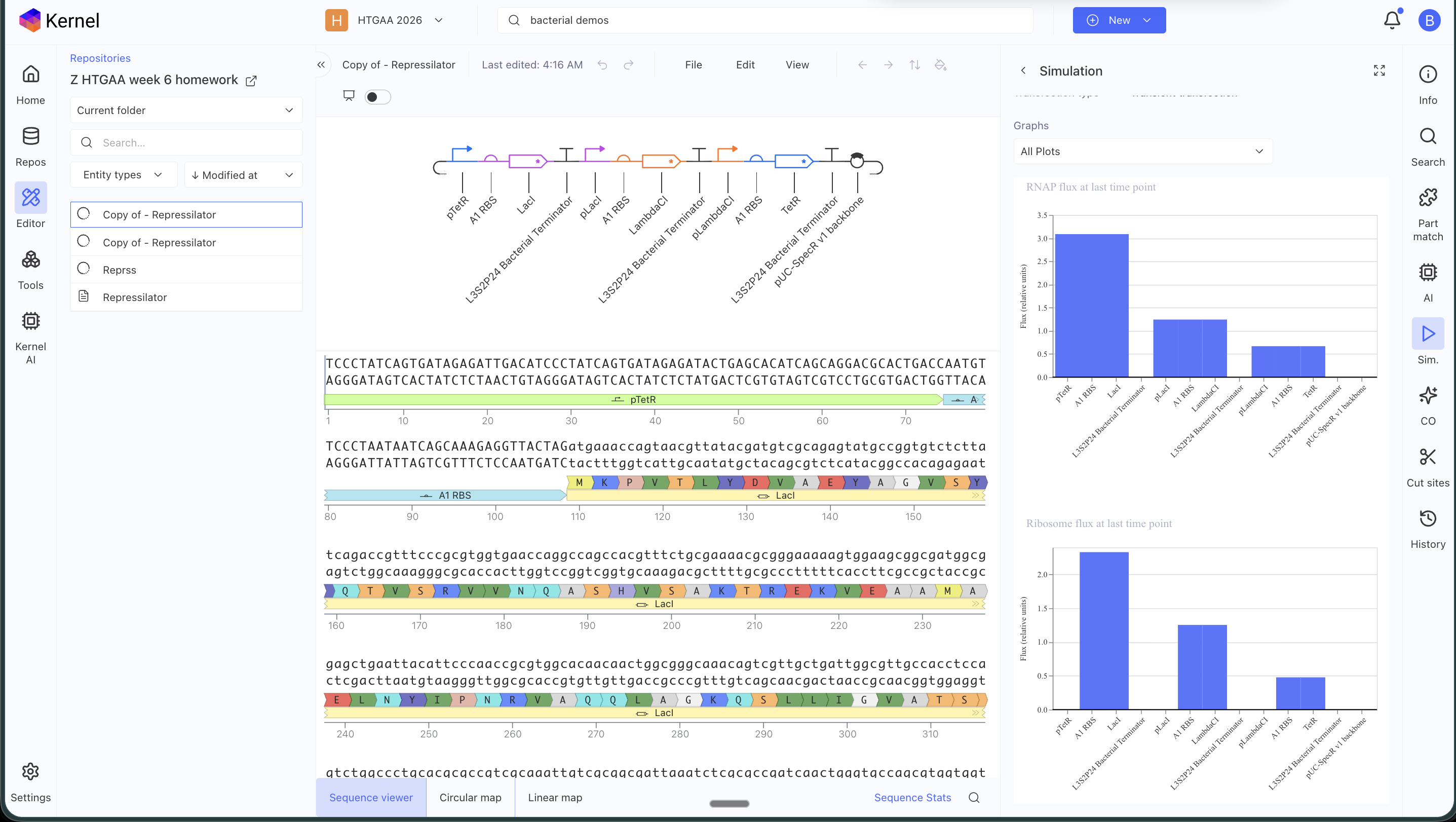
My simulation graphs did not match:
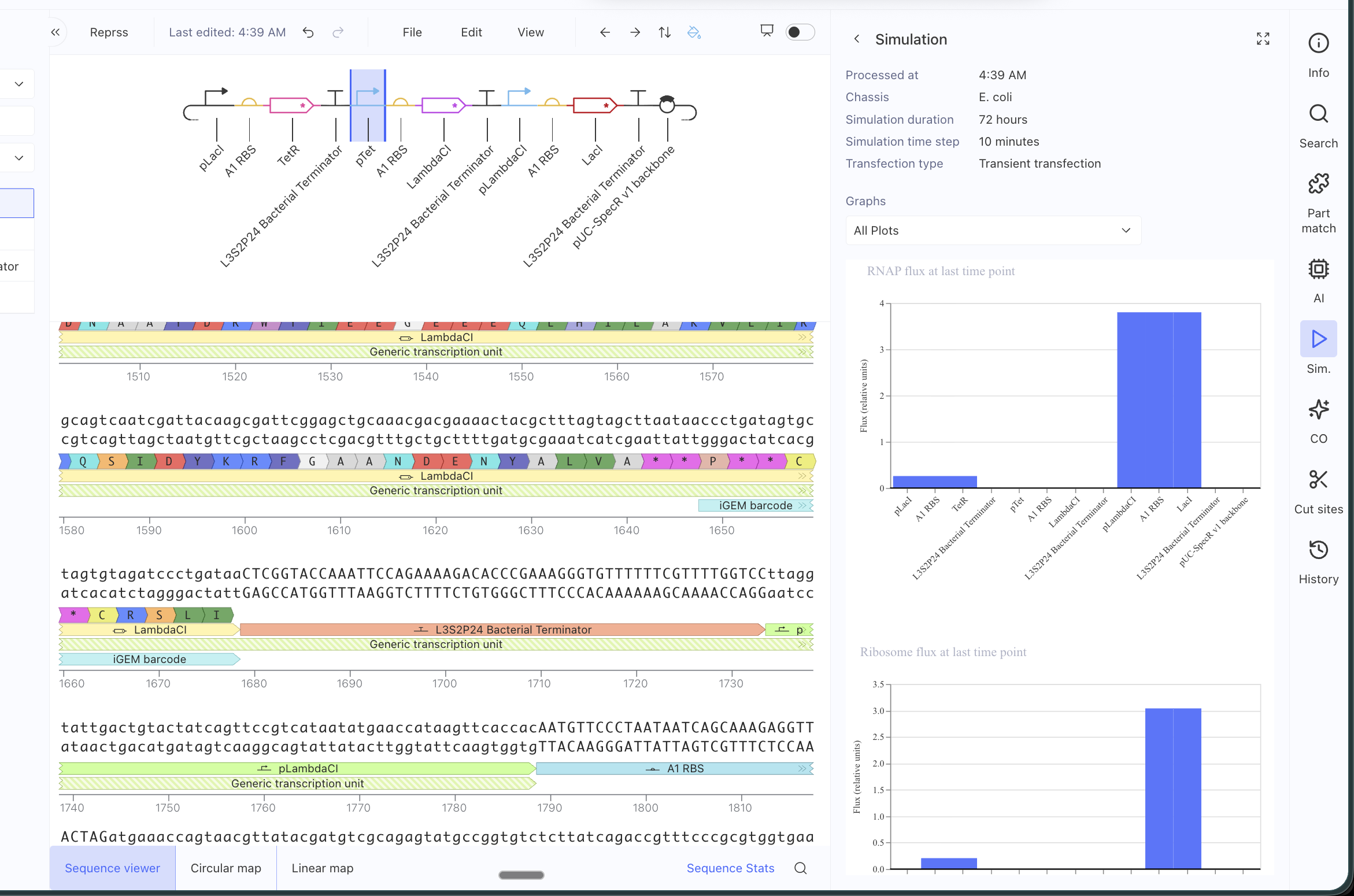 Even after replacing all the parts specifically for the ones in the Characterized Bacteria repo, it still didn’t match. I will have to debug further.
Even after replacing all the parts specifically for the ones in the Characterized Bacteria repo, it still didn’t match. I will have to debug further.
I asked AI to help me debug and it said that some possible reasons might be that the parts I am using might have different metadata even if they appear identical, that my simulation window might be too long, that the RBS I chose might be too weak or strong, that the backbone or copy-number might differ (I tried with and without an added backbone), that the promoters might not have the right repressor behavior… many alternatives to test!