Class assignment – designing policy frameworks around bioengineering tools ⭐⭐ Web app with policy homework content ⭐⭐ (external link)
🔗✏️ Proof-of-work links (drafts, AI conversations/prompts used to brainstorm, generate the web app, and other sections) ✏️🔗
HTGAA 2026: Week 1 Governance Framework Project: DIY Aging Biomarkers Kit for Community Labs
Objective: To lower the single biggest barrier to longevity research (cost) through an affordable, open-source testing kit.
PART 2 path to image: 2026a-zoe-isabel-senon/webpages/src/branch/main/assets/images/heartsim.jpg
(I tried to recreate a heart shape…)
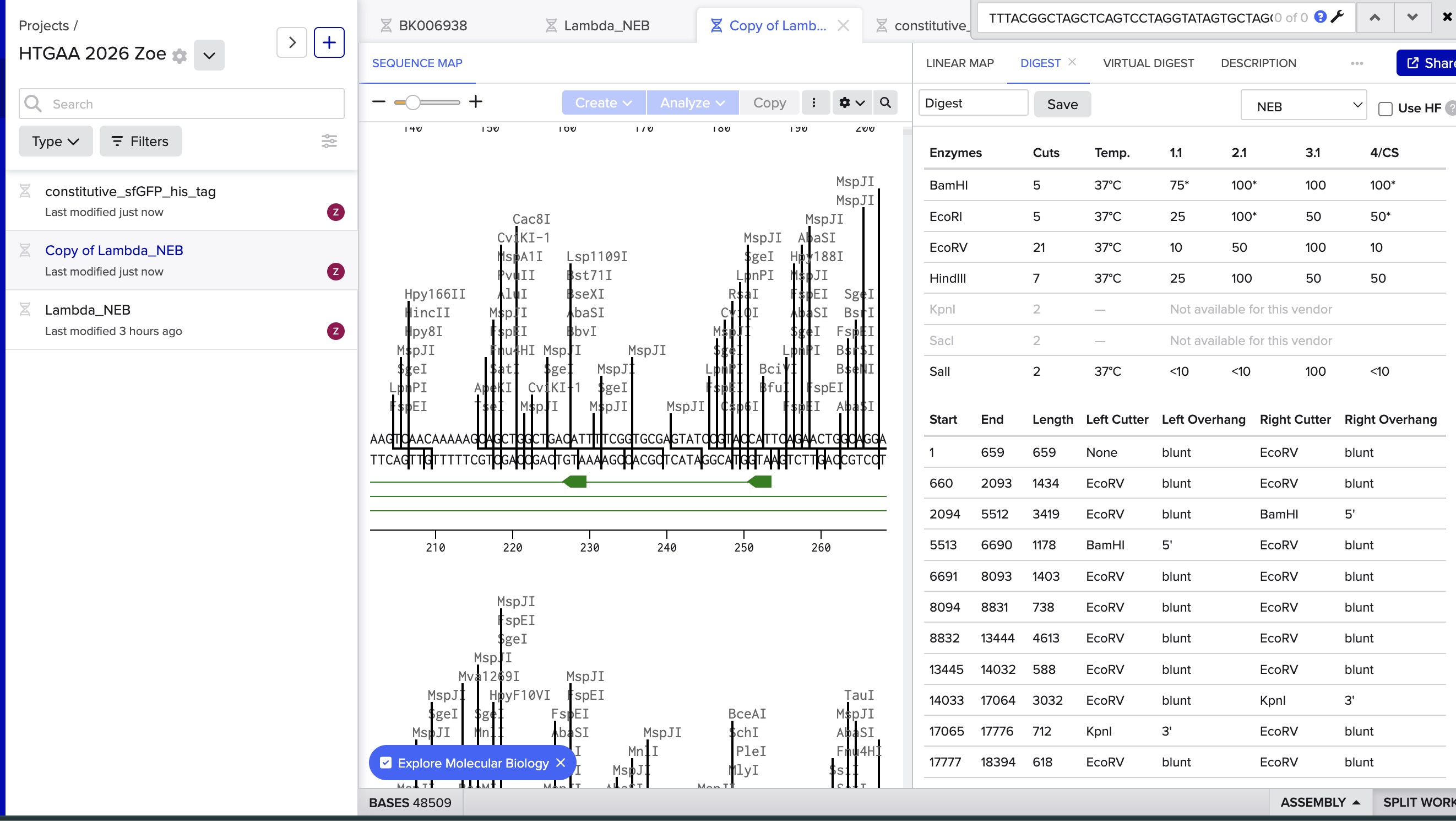
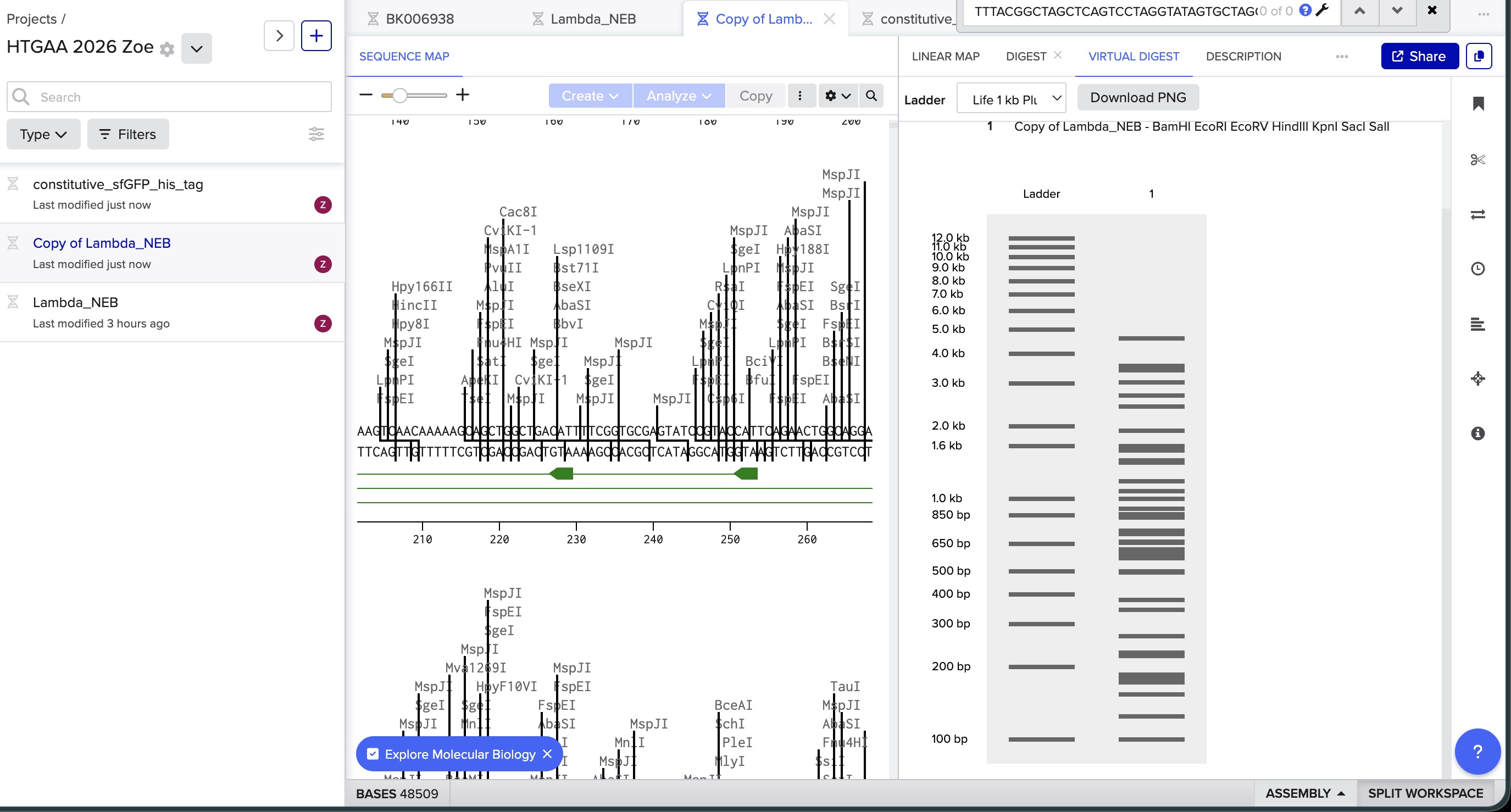
I did the digest simulation in Benchling and checked the virtual gel simulation result:
image in: 2026a-zoe-isabel-senon/webpages/src/branch/main/assets/images/benchlingdigest1.jpg
image in: 2026a-zoe-isabel-senon/webpages/src/branch/main/assets/images/benchlingdigest2.jpg
3.1 PICK A PROTEIN & Find the protein sequence: I picked SIRT1
⛓️ Collab Notebook Link ⛓️ Link to the Colab for the chosen protein (FOXO3): Collab notebook
💡 PART A - CONCEPTUAL QUESTIONS How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
🔎 Part 1: Generate Binders with PepMLM Sequence with mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
![[htgaa-hw5-pt1-binders.png]]
In text, the generated ones (plus the one provided to compare) were:
0 WRYYAAALRHKX 9.957530 1 WRYYAVAARHKK 13.948671 2 WRSYVVVLELGX 18.185468 3 HHYPAVAVALKX 7.987091 4 FLYRWLPSRRGG 20.635231 AlphaFold however was throwing errors with these sequences, so I asked Claude to check: ![[Screenshot 2026-03-08 at 17.05.41.png]]
SECTION 1: PCR & GIBSON ASSEMBLY “What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? It is composed of 3 essential components: 1. Phusion DNA Polymerase: a high-fidelity enzyme with low error rates (50x higher fidelity than Taq) and fast extension speeds (15-30sec/kilobase) 2. dNTPs (deoxynucleotide triphosates) (dATP, dCTP,dGTP, dTTP): the nucleotide building blocks the polymerase incorporates into the DNA starnd it builds 3. An optimized reaction buffer containing Mg(^2+) ions (polymerase cofactor that also stabilizes annealing) and KCl, which promotes specific primer binding while suppressing non-specific interactions. The Green Master Mix also contains other reagents (green dye, density reagents) so the reaction can be directly loaded into an agarose gel.
PART 0 1. How do ERNs work + how this differs from proteases They cleave mRNA transcripts before they can be translated into proteins, so the mRNA is not processed by the ribosomes and gets degraded quickly by cellular exonucleases. They work directly at the RNA level, unlike proteases which work on the final translated proteins. This means that they prevent new proteins from being produced, but they do not affect the existing pool of proteins.
Week 9 Homework: Cell-Free Systems [WIP!]
Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. The benefit can be summarized as “it removes the constraints we usually face for protein synthesis when working with live cells”. For example, working with live cells requires culturing cells throughout the whole cell lifecycle, with all the possibilities for error that this implies (mistakes, unexpected cell behavior due to their non-deterministic nature, etc), as well as the required timelines (speed is limited by the fundamental speed constraints from the cell growth cycle), and costs. Cell-free systems also allow for much greater control, as the main system is boiled down to its most basic functional components, removing a lot of complexity (variables outside of our control), and therefore allowing the possibility of producing much more homogeneous products.
Project: DIY Aging Biomarkers Kit for Community Labs Objective: To lower the single biggest barrier to longevity research (cost) through an affordable, open-source testing kit.
Context: Diseases of aging account for 3/4 of global deaths, yet independent research is gated by prohibitive costs (often $500+ per commercial assay).
Proposed Tool: An accessible, open-source DIY Aging Biomarkers Kit designed specifically for the ~50 community biolabs worldwide. This kit allows independent researchers to measure cellular senescence and other aging hallmarks without relying on expensive institutional supply chains.
*(Bonus: Personal context for why I chose this project 🙋🏼♀️)*
I am extremely passionate about the topic of “solving aging” as a way to reduce suffering in the world at scale (as typically called “diseases of aging” ultimately account for somewhere between 1/3 and ~3/4 of all deaths (1) (2)).
With this goal in mind, I decided to fully focus my efforts on biotech, coming from a computational background, and learn hands-on by joining an open community lab (Biopunk Labs in San Francisco – one of the HTGAA nodes) and running my own bioengineering experiments.
For my second experiment I decided to use mammalian cells (fibroblasts) instead of bacteria, as bacteria do not share our mechanisms of aging and are therefore not a good model to study this.
However, I quickly realized how severely limited the scope of what I could do in the lab would be due to the high costs associated with working independently in the lab (without external funding), especially when mammalian cells are involved. Initially, I was planning to replicate a full cellular reprogramming protocol, but quickly learned this would not be a possibility for me due to the cost, so I pivoted to a “partial reprogramming” protocol.
I managed to secure sample cells and some media through donations, thanks to some very kind fellow scientists and mentors who wanted to support such an initiative*. However, none of my work would have any validity unless I could find a reliable way to measure the effects of my interventions on biomarkers classically associated with longevity (see: Hallmarks of Aging), such as senescence.
*(people thought I was crazy for even attempting to run mammalian cell experiments independently, as a beginner, given the costs and difficulty!)
I quickly learned that assay kits would quickly eat up a big portion of my budget, especially given that most companies sell them in bulk, which makes them prohibitively expensive for independent researchers wanting to run just a few small assays.
This whole experience opened my eyes to how non-accessible aging research is at moment.
Given that a big part of my mission is to enable others to help accelerate the eradication of all human disease (with a special focus on longevity), a straightforward way in which I could contribute to this acceleration is by enabling more independent research to happen by lowering the biggest entry barrier: the cost to even get started with simple longevity experiments.
02. Policy Objectives
Democratize Access: Lower financial and institutional barriers so scientists in low-resource settings can meaningfully contribute to aging research.
Prevent Misuse: Ensure that wider access to bioengineering tools does not enable unsafe practices through proactive monitoring.
Ensure Translation: Guarantee that research outcomes meet quality standards sufficient for peer review and clinical translation.
03. Proposed Interventions
A1: Government Subsidies (Incentive)
Actor: Government funding bodies (NIH, ERC) A dedicated micro-grant program (€5–25K) for community labs to accelerate biomedical progress by enabling new and existing scientists to carry out independent aging research.
Risk: Could create a two-tier system where funded labs professionalize and lose grassroots accessibility.
A2: Lab Space Sharing Network (Coordination)
Actor: Academic Institutions, Incubators An “Airbnb for lab benches” connecting institutions with spare capacity to independent researchers.
Risk: Institutions may refuse due to liability concerns.
A3: AI Biosafety Co-Pilot (Technical)
Actor: Open-source developers, DIYbio.org A software tool that checks experiment plans against biosafety databases, flags risks, and requires mentor sign-off. Serves as a dynamic guardrail.
Risk: Over-reliance could reduce researchers’ own safety judgment (the “GPS effect”).
04. Impact Assessment Matrix
Criteria
A1: Subsidies
A2: Lab Network
A3: AI Co-Pilot
Access Democratization
High
High
Indirect
Enables new talent
✓
✓
✓
Enables existing researchers
✓
✓
—
Misuse Prevention
—
—
Most
Ensures safety standards
—
—
✓
Blocks malign applications
—
—
✓
Translation
Partial
Partial
—
Incentivizes translation
✓
✓
—
Directly aids translation
✓
✓
—
Feasibility
Low
Medium
High
Legend: ✓ = Strong positive contribution — = Not applicable / No direct impact
Assignment (Week 2 Lecture Prep)
From Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Polymerase’s error rate is 1:106 (one error per 106 bases), about one in ten million. The human genome is 3.2 Gbp (3.2 * 109), which would then mean that we would expect to see 320 errors per haploid genome copy with no additional repair mechanisms (or double that amount for diploid replication). Yet we see that the actual final mutation rate is closer to 1 in 109, which is multiple orders of magnitude less.
This is because the body applies additional “proofreading” and repair mechanisms, such as by leveraging mismatch repair proteins that scan newly synthesized DNA (they can recognize helix distortions where bases are mismatched and resynthesize them). Additionally, polymerase itself has a 3’ to 5’ exonuclease activity that removes incorrectly incorporated nucleotides immediately, allowing polymerase to redo the badly-synthesized base section.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice, what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Given that an average human protein is encoded by 1036 bp, and each codon consists of 3 bases, we can calculate that each protein is approximately 345 amino acids long (1036/3 ≈345).
Since there are 61 codons that can code for 20 amino acids (64 total codons, but 3 of them are “stop” codons), and multiple codons can code for the same amino acid, a theoretically enormous number of DNA sequences could code for the same protein. Yet in practice we see that only a limited set of codon combinations actually lead to the production of a specific protein, and this is because there are additional biological limitations that dictate these capabilities: certain combinations are more viable (or not viable at all) based on external factors like the availability of matching tRNAs to produce it, or a given organism’s preference for a specific codon sequence, regulatory sequences that might be accidentally created, mRNA secondary structure stability, and translation efficiency.
From Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Why can’t you make a 2000bp gene via direct oligo synthesis?
The current standard for de novo gene synthesis is solid‑phase phosphoramidite chemistry. It is effective but highly noisy (errors accumulate rapidly on each iteration due to approximately 95-99.5% stepwise coupling efficiency), which currently limits the total amount of viable DNA that can be synthesized, as, for a coding gene, a single base error can cause malfunction.
200 nt is the standard limit because at that point most strands have some error, but at a level where it is still feasible to fix it through cloning or purification.
A 2,000 bp gene would require 2,000 single-stranded nucleotides. The most optimistic coupling efficiency per base (99.9%), the accumulation of errors (only about 13.5% of synthesized strands would be full-length and error-free) would make the final product differ too much from the desired sequence to be useful.
We can, however, assemble genes of lengths much longer than the theoretical limitation for the method by synthesizing much smaller and more manageable strands (~50-150 bp) and assembling them through methods like PCR and Gibson assembly. This is the standard for longer DNA synthesis.
From George Church
Using Google & Prof. Church’s slide #4: What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids are
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Threonine
Tryptophan
Valine
Arginine
(Note: in humans, arginine is considered conditionally essential and is sometimes included in the list of 10)
The definition of an “essential amino acid” is that it must be obtained from external sources because the organism cannot produce it on its own, so by this definition, and given that lysine is already found in this category, the “lysine contingency” would be a nonsensical solution (even without additional bioengineering, the dinosaurs would still have to rely on obtaining lysine from external sources like lysine-rich prey and plants, and in the movie they are already shown consuming these).
Week 2 HW: DNA Sequencing and Synthesis
PART 2
path to image: 2026a-zoe-isabel-senon/webpages/src/branch/main/assets/images/heartsim.jpg
(I tried to recreate a heart shape…)
I did the digest simulation in Benchling and checked the virtual gel simulation result:
Obtained through the reverse translation tool after attaching the human codon table:
```jsx
Reverse Translate
Results for 747 residue sequence "sp|Q96EB6|SIR1_HUMAN NAD-dependent protein deacetylase sirtuin-1 OS=Homo sapiens OX=9606 GN=SIRT1 PE=1 SV=2" starting "MADEAALALQ"
>reverse translation of sp|Q96EB6|SIR1_HUMAN NAD-dependent protein deacetylase sirtuin-1 OS=Homo sapiens OX=9606 GN=SIRT1 PE=1 SV=2 to a 2241 base sequence of most likely codons.
atggccgacgaggccgccctggccctgcagcccggcggcagccccagcgccgccggcgcc
gacagagaggccgccagcagccccgccggcgagcccctgagaaagagacccagaagagac
ggccccggcctggagagaagccccggcgagcccggcggcgccgcccccgagagagaggtg
cccgccgccgccagaggctgccccggcgccgccgccgccgccctgtggagagaggccgag
gccgaggccgccgccgccggcggcgagcaggaggcccaggccaccgccgccgccggcgag
ggcgacaacggccccggcctgcagggccccagcagagagccccccctggccgacaacctg
tacgacgaggacgacgacgacgagggcgaggaggaggaggaggccgccgccgccgccatc
ggctacagagacaacctgctgttcggcgacgagatcatcaccaacggcttccacagctgc
gagagcgacgaggaggacagagccagccacgccagcagcagcgactggacccccagaccc
agaatcggcccctacaccttcgtgcagcagcacctgatgatcggcaccgaccccagaacc
atcctgaaggacctgctgcccgagaccatccccccccccgagctggacgacatgaccctg
tggcagatcgtgatcaacatcctgagcgagccccccaagagaaagaagagaaaggacatc
aacaccatcgaggacgccgtgaagctgctgcaggagtgcaagaagatcatcgtgctgacc
ggcgccggcgtgagcgtgagctgcggcatccccgacttcagaagcagagacggcatctac
gccagactggccgtggacttccccgacctgcccgacccccaggccatgttcgacatcgag
tacttcagaaaggaccccagacccttcttcaagttcgccaaggagatctaccccggccag
ttccagcccagcctgtgccacaagttcatcgccctgagcgacaaggagggcaagctgctg
agaaactacacccagaacatcgacaccctggagcaggtggccggcatccagagaatcatc
cagtgccacggcagcttcgccaccgccagctgcctgatctgcaagtacaaggtggactgc
gaggccgtgagaggcgacatcttcaaccaggtggtgcccagatgccccagatgccccgcc
gacgagcccctggccatcatgaagcccgagatcgtgttcttcggcgagaacctgcccgag
cagttccacagagccatgaagtacgacaaggacgaggtggacctgctgatcgtgatcggc
agcagcctgaaggtgagacccgtggccctgatccccagcagcatcccccacgaggtgccc
cagatcctgatcaacagagagcccctgccccacctgcacttcgacgtggagctgctgggc
gactgcgacgtgatcatcaacgagctgtgccacagactgggcggcgagtacgccaagctg
tgctgcaaccccgtgaagctgagcgagatcaccgagaagccccccagaacccagaaggag
ctggcctacctgagcgagctgccccccacccccctgcacgtgagcgaggacagcagcagc
cccgagagaaccagcccccccgacagcagcgtgatcgtgaccctgctggaccaggccgcc
aagagcaacgacgacctggacgtgagcgagagcaagggctgcatggaggagaagccccag
gaggtgcagaccagcagaaacgtggagagcatcgccgagcagatggagaaccccgacctg
aagaacgtgggcagcagcaccggcgagaagaacgagagaaccagcgtggccggcaccgtg
agaaagtgctggcccaacagagtggccaaggagcagatcagcagaagactggacggcaac
cagtacctgttcctgccccccaacagatacatcttccacggcgccgaggtgtacagcgac
agcgaggacgacgtgctgagcagcagcagctgcggcagcaacagcgacagcggcacctgc
cagagccccagcctggaggagcccatggaggacgagagcgagatcgaggagttctacaac
ggcctggaggacgagcccgacgtgcccgagagagccggcggcgccggcttcggcaccgac
ggcgacgaccaggaggccatcaacgaggccatcagcgtgaagcaggaggtgaccgacatg
aactaccccagcaacaagagc
>reverse translation of sp|Q96EB6|SIR1_HUMAN NAD-dependent protein deacetylase sirtuin-1 OS=Homo sapiens OX=9606 GN=SIRT1 PE=1 SV=2 to a 2241 base sequence of consensus codons.
atggcngaygargcngcnytngcnytncarccnggnggnwsnccnwsngcngcnggngcn
gaymgngargcngcnwsnwsnccngcnggngarccnytnmgnaarmgnccnmgnmgngay
ggnccnggnytngarmgnwsnccnggngarccnggnggngcngcnccngarmgngargtn
ccngcngcngcnmgnggntgyccnggngcngcngcngcngcnytntggmgngargcngar
gcngargcngcngcngcnggnggngarcargargcncargcnacngcngcngcnggngar
ggngayaayggnccnggnytncarggnccnwsnmgngarccnccnytngcngayaayytn
taygaygargaygaygaygaygarggngargargargargargcngcngcngcngcnath
ggntaymgngayaayytnytnttyggngaygarathathacnaayggnttycaywsntgy
garwsngaygargargaymgngcnwsncaygcnwsnwsnwsngaytggacnccnmgnccn
mgnathggnccntayacnttygtncarcarcayytnatgathggnacngayccnmgnacn
athytnaargayytnytnccngaracnathccnccnccngarytngaygayatgacnytn
tggcarathgtnathaayathytnwsngarccnccnaarmgnaaraarmgnaargayath
aayacnathgargaygcngtnaarytnytncargartgyaaraarathathgtnytnacn
ggngcnggngtnwsngtnwsntgyggnathccngayttymgnwsnmgngayggnathtay
gcnmgnytngcngtngayttyccngayytnccngayccncargcnatgttygayathgar
tayttymgnaargayccnmgnccnttyttyaarttygcnaargarathtayccnggncar
ttycarccnwsnytntgycayaarttyathgcnytnwsngayaargarggnaarytnytn
mgnaaytayacncaraayathgayacnytngarcargtngcnggnathcarmgnathath
cartgycayggnwsnttygcnacngcnwsntgyytnathtgyaartayaargtngaytgy
gargcngtnmgnggngayathttyaaycargtngtnccnmgntgyccnmgntgyccngcn
gaygarccnytngcnathatgaarccngarathgtnttyttyggngaraayytnccngar
carttycaymgngcnatgaartaygayaargaygargtngayytnytnathgtnathggn
wsnwsnytnaargtnmgnccngtngcnytnathccnwsnwsnathccncaygargtnccn
carathytnathaaymgngarccnytnccncayytncayttygaygtngarytnytnggn
gaytgygaygtnathathaaygarytntgycaymgnytnggnggngartaygcnaarytn
tgytgyaayccngtnaarytnwsngarathacngaraarccnccnmgnacncaraargar
ytngcntayytnwsngarytnccnccnacnccnytncaygtnwsngargaywsnwsnwsn
ccngarmgnacnwsnccnccngaywsnwsngtnathgtnacnytnytngaycargcngcn
aarwsnaaygaygayytngaygtnwsngarwsnaarggntgyatggargaraarccncar
gargtncaracnwsnmgnaaygtngarwsnathgcngarcaratggaraayccngayytn
aaraaygtnggnwsnwsnacnggngaraaraaygarmgnacnwsngtngcnggnacngtn
mgnaartgytggccnaaymgngtngcnaargarcarathwsnmgnmgnytngayggnaay
cartayytnttyytnccnccnaaymgntayathttycayggngcngargtntaywsngay
wsngargaygaygtnytnwsnwsnwsnwsntgyggnwsnaaywsngaywsnggnacntgy
carwsnccnwsnytngargarccnatggargaygarwsngarathgargarttytayaay
ggnytngargaygarccngaygtnccngarmgngcnggnggngcnggnttyggnacngay
ggngaygaycargargcnathaaygargcnathwsngtnaarcargargtnacngayatg
aaytayccnwsnaayaarwsn
```
3.3 - In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
I have chosen to optimize it for homo sapiens, because I am interested in human medicine and I want to learn more about how certain proteins might affect human lifespans.
We need to optimize codon usage because different organisms have different availability of tRNA to match certain codons, and we want to optimize for tRNA abundance to maximize translation speed, and mRNA stability and structure. These optimizations can be made because the same amino acid can be produced with different codons.
3.4 - What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
I could transfect the necessary genes into mammalian cells (like neurons, or HEK cells, which divide rapidly!) to make them express this protein in large quantities. We do this by basically integrating these “instructions” (as, for example, plasmids) for producing different specific proteins into the cell, and letting their machinery decode them in the same way that it does with their own genome.
(I have done this in the past with HEK cells to make them express RFP, using lipofectamine as the transfection method/reagent.)
We optimize the plasmid content for the target organism by choosing a certain promoter, codon-optimized CDS, and regulatory elements.
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would want to sequence the genomes of healthy centenarians or supercentenarians (people who reach 110 years of age), so that we can learn if there is a genetic basis for longevity and find enhancing variants. I know that certain variants of the FOXO3 gene have been found to be more prevalently enriched among this population (paper), and I would like to learn more.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?**
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.**
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?**
What is the output of your chosen sequencing technology?**
I would choose Illumina short-read WGS, which is a relatively quick, affordable and accurate current industry standard for consumer sequencing.
It is a second-generation method that allows to do massively-parallelized short-reads.
To do this, we would take a sample from the supercentenarians (saliva or blood), perform end-repair and A-tailing, ligate Illumina adapters, and PCR-amplify to make libraries.
The output is a FASTQ file which we align to the human reference genome to find (call) SNPs, indels, etc. Basically to identify how and where this given genome differs from the “reference” one (assembled as an average of a group of +100 individuals).
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?**
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?**
I would use the industry standard (phosphoramidite chemical oligo synthesis + Gibson assembly) as it is currently the most battle-tested one.
The limitation is that it only allows us to assemble up to around 200 nucleotides at a time before the error rate becomes so high as to make the output unusable (too different from the referent we want to “print”). To print out longer sequences, we must separately assemble them using methods like Gibson assembly.
5.3 DNA Edit
(i) What DNA would you want to edit and why? What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Genes associated with longevity or cognitive enhancement, like FOXO3 or Klotho. I would like to test these in both humans and animals, especially dogs, since it could be helpful to obtain faster data about the effects in mammals, and possibly buy a few people additional time with their beloved pets :)
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?**
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?**
What are the limitations of your editing methods (if any) in terms of efficiency or precision?**
I would use a CRISPR-based editing system and deliver it with AAV (the current most widely-used delivery mechanism for gene therapies). AAV is safer because it doesn’t integrate, which means that it doesn’t actually modify the existing genome (which can cause issues like cancer through insertional mutagenesis).
The main limitation of AAVs is their small cargo sizes, which severely limits the number of genes we could deliver through this method.
Why this paper:
This paper presents an open-source system built around the Opentrons OT-2 that automates the passaging of adherent or suspension cells in 96-well plates. What makes it novel is the integration of a custom Cell Counting Imager (CCI) that adjusts seeding volumes per-well based on measured cell density (the robot doesn’t just pipette blindly, it makes decisions based on real-time data). The system fits inside a biosafety cabinet, processes 2–3 plates per working day with minimal intervention, and communicates status updates via Slack.
Relevance to my final project ideas:
All three of my proposed projects involve cell culture steps. Automated cell passaging with integrated counting is exactly the infrastructure needed upstream of any of these experiments. The paper also demonstrates how custom hardware (the CCI) can be integrated with the OT-2’s Python API, which is relevant for designing my own automation protocols that might need plate reader integration for fluorescence readouts.
⚙️ Assignment: How I’ll apply automation in my projects
This is how I’d use automation for my proposed final project #1 (“Idea 1: CRISPR Epigenome Editing at Clock CpG Sites”):
Step 1 — Transfection Setup (Opentrons OT-2/Flex)
Dispensing cells, transfection reagents (e.g., Lipofectamine or PEI), and different gRNA/construct combinations across a 96-well plate. Each well gets a different gRNA targeting a different CpG site, allowing parallel testing of multiple epigenome editing targets.
Step 2 — Bisulfite Conversion Sample Prep (Opentrons OT-2)
After editing, validate whether methylation was actually changed at the target site. This requires bisulfite conversion of genomic DNA — a multi-step protocol (denaturation → bisulfite treatment → desulphonation → cleanup) that is repetitive, error-prone, and ideal for automation. Zymo Research already provides Opentrons-compatible scripts for their EZ DNA Methylation kits.
Step 3 — Targeted PCR Setup (Opentrons OT-2)
Set up PCR reactions targeting the specific CpG sites of interest (using bisulfite-specific primers) across the 96-well plate for downstream sequencing.
Pseudocode for Transfection Protocol:
from opentrons import protocol_api
metadata = {
'protocolName': 'dCas9-TET1 Epigenome Editing Transfection Setup',
'author': '[Your Name]',
'description': 'Automated transfection of dCas9-TET1 + gRNAs targeting aging clock CpG sites'
}
requirements = {"robotType": "OT-2", "apiLevel": "2.16"}
def run(protocol: protocol_api.ProtocolContext):
# --- LABWARE ---
plate = protocol.load_labware('corning_96_wellplate_360ul_flat', '1')
reagent_rack = protocol.load_labware('opentrons_24_tuberack_nest_1.5ml', '2')
tiprack_20 = protocol.load_labware('opentrons_96_tiprack_20ul', '3')
tiprack_300 = protocol.load_labware('opentrons_96_tiprack_300ul', '6')
# --- INSTRUMENTS ---
p20 = protocol.load_instrument('p20_single_gen2', 'left', tip_racks=[tiprack_20])
p300 = protocol.load_instrument('p300_single_gen2', 'right', tip_racks=[tiprack_300])
# --- REAGENTS (in tube rack) ---
# A1: OptiMEM reduced serum media
# A2: Lipofectamine 3000 reagent
# A3: P3000 reagent
# A4: dCas9-TET1 plasmid (shared across all wells)
# B1-B6: gRNAs targeting 6 different aging clock CpG sites
# B1: gRNA for ELOVL2 promoter (cg16867657)
# B2: gRNA for FHL2 promoter (cg22454769)
# B3: gRNA for PENK intron (cg16008966)
# B4: gRNA for KLF14 promoter (cg07553761)
# B5: gRNA for TRIM59 (cg00481951)
# B6: non-targeting control gRNA
# C1: Cell suspension (pre-trypsinized, counted)
optimen = reagent_rack['A1']
lipofectamine = reagent_rack['A2']
p3000 = reagent_rack['A3']
dcas9_tet1 = reagent_rack['A4']
grnas = [reagent_rack[well] for well in ['B1', 'B2', 'B3', 'B4', 'B5', 'B6']]
cells = reagent_rack['C1']
# --- PROTOCOL ---
# Step 1: Seed cells into all wells (200 uL per well, ~40,000 cells)
# Note: In practice, cells would be seeded 24h prior
# This step shows the protocol for Day 0
protocol.comment("Step 1: Seeding cells")
for well in plate.wells()[:48]: # 48 wells = 6 gRNAs x 8 replicates
p300.transfer(200, cells, well, new_tip='always')
# [Protocol would pause here for 24h incubation]
protocol.pause("Incubate plate at 37°C, 5% CO2 for 24 hours. Resume for transfection.")
# Step 2: Prepare transfection complexes
# Each gRNA condition gets 8 replicate wells (columns)
protocol.comment("Step 2: Preparing lipofection complexes")
for grna_idx, grna in enumerate(grnas):
# Target wells: 8 wells per gRNA (one column)
target_wells = plate.columns()[grna_idx][:8]
for well in target_wells:
# Add OptiMEM
p20.transfer(10, optimen, well, new_tip='always')
# Add dCas9-TET1 plasmid (250 ng)
p20.transfer(2, dcas9_tet1, well, new_tip='always')
# Add specific gRNA plasmid (250 ng)
p20.transfer(2, grna, well, new_tip='always')
# Add P3000 reagent
p20.transfer(1, p3000, well, new_tip='always')
# Add Lipofectamine
p20.transfer(1, lipofectamine, well, mix_after=(3, 10), new_tip='always')
protocol.comment("Transfection complete. Incubate 72h before harvesting.")
# After 72h: harvest cells for bisulfite conversion + methylation analysis
Pseudocode for Bisulfite Conversion (Post-editing validation):
def run_bisulfite_conversion(protocol: protocol_api.ProtocolContext):
"""
Automated bisulfite conversion of genomic DNA from transfected cells.
Validates whether dCas9-TET1 successfully demethylated target CpG sites.
Uses Zymo EZ DNA Methylation Kit protocol adapted for OT-2.
"""
# Step 1: Add CT Conversion Reagent to each DNA sample
# 130 uL CT reagent + 20 uL DNA sample per well
for well in sample_plate.wells()[:48]:
p300.transfer(130, ct_reagent, well, mix_after=(5, 100))
# Step 2: Thermal cycling for bisulfite conversion
# Would use Opentrons Thermocycler Module:
# 98°C for 8 min (denaturation)
# 64°C for 3.5 hours (conversion)
# 4°C hold
thermocycler.set_block_temperature(98, hold_time_minutes=8)
thermocycler.set_block_temperature(64, hold_time_minutes=210)
thermocycler.set_block_temperature(4)
# Step 3: Desulphonation + Cleanup
# Transfer to spin columns, wash, elute
# (Magnetic bead-based cleanup would be more OT-2 friendly)
for well in sample_plate.wells()[:48]:
# Add binding buffer
p300.transfer(600, binding_buffer, well, mix_after=(3, 200))
# Transfer to magnetic bead plate
# Wash 2x with wash buffer
# Elute in 10 uL elution buffer
p20.transfer(10, elution_buffer, well, mix_after=(5, 8))
# Step 4: Set up targeted PCR for clock CpG sites
# Bisulfite-specific primers for each target locus
for i, primer_pair in enumerate(bisulfite_primers):
target_wells = pcr_plate.columns()[i][:8]
for well in target_wells:
p20.transfer(10, pcr_master_mix, well)
p20.transfer(2, primer_pair, well)
p20.transfer(8, converted_dna, well, mix_after=(3, 15))
💡 Final project ideas
Added the slides to the slide deck.
3 final project ideas I’d like to pursue
Design targeted epigenetic editors to reverse aging methylation signatures
Training a Minimal Epigenetic Aging Clock and Design of a Fluorescent Age Reporter Construct
Cross-Intervention Epigenetic Clock Comparison and CRISPR Knock-in Design of a p16 Aging Sensor
Week 4 HW: Protein Design Part I
⛓️ Collab Notebook Link
⛓️ Link to the Colab for the chosen protein (FOXO3): Collab notebook
💡 PART A - CONCEPTUAL QUESTIONS
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Avogadro’s number: 1 mole of anything = 6.022 * 10^23 molecules. Since 1 Da = 1 g/mol, a molecule with mass 100 Da has a molar mass of 100 g/mol.
so: 500g / (100g/mol) = 5 mo * 6.022*(10^23) = 3.01e+24 molecules
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The cells and proteins contained in the food are completely broken down into their most essential forms (ie amino acids) before being absorbed and used by the body.
The proteins contained in the food are not used directly, instead they are broken down into amino acids which are then used as building materials when our cellular machine reads our own DNA to build the proteins we need (which might be different than the versions encoded by other species).
Why are there only 20 natural amino acids?
A “why” question can never be rigurously answered in biology, but we can infer that this set of amino acids contains all the necessary properties to create life.
(Note: by doing this exercise, I learned that there are actually 22 amino acids in nature, with the 2 additional ones being selenocystein and pyrrolysine)
Can you make other non-natural amino acids? Design some new amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
In 1953, scientists Stanley Miller and Harold Urey were able to produce amino acids by simulating early Earth conditions, by using electric sparks in a mixture of water, ammonia, methane and hydrogen.
Other possible sources are meteorites (which have been found to be rich in amino acids), hydrothermal vents, photochemistry (UV radiation on ice surfaces) and, potentially, Strecker synthesis.
If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed. They are the mirror image of L-amino acids, which are right-handed for the following reason:
”Natural L-amino acids have their φ (phi) and ψ (psi) backbone dihedral angles in a region of the Ramachandran plot (approximately φ = −60°, ψ = −40°) that produces a right-handed helix”
((Curious fact: origin of the “D” and “L” I just learned)) (nested/click to see)
“The letters L and D come from an old naming system that compares molecules to the simple sugar glyceraldehyde and to “left/right” in Latin.
Historical origin: Chemists first defined D‑ and L‑ using glyceraldehyde: in a standard Fischer projection, the isomer with the key group on the right was called D (from dextro = right), and the one with it on the left was called L (from laevo = left).
Can you discover additional helices in proteins?
Yes, I learned that there’s other types like n-helix, collagen triple helix,
Why are most molecular helices right-handed?
Why do β-sheets tend to aggregate?
Because the edge strands of every β-sheet have unsatisfied hydrogen bond capacity, and are therefore “looking” for more strands to bond with.
- What is the driving force for β-sheet aggregation?
The primary driver is hydrogen bonding. Each hydrogen bond contributes ~2-8kJ/mol, so a sheet with many bonds has a large energetic benefit.
Why do many amyloid diseases form β-sheets?
”Amyloids are ordered protein aggregates with a characteristic cross-β structure: β-strands run perpendicular to the fibril long axis, forming a “spine” of stacked sheets. They are extraordinarily stable: resistant to heat, most detergents, and proteases.”
- Can you use amyloid β-sheets as materials?
-
Yes. Some examples I have found: spider silk, Curli fibers, biotemplating for metallic wires.
13. Design a β-sheet motif that forms a well-ordered structure.
💡 Part B: Protein Analysis and Visualization
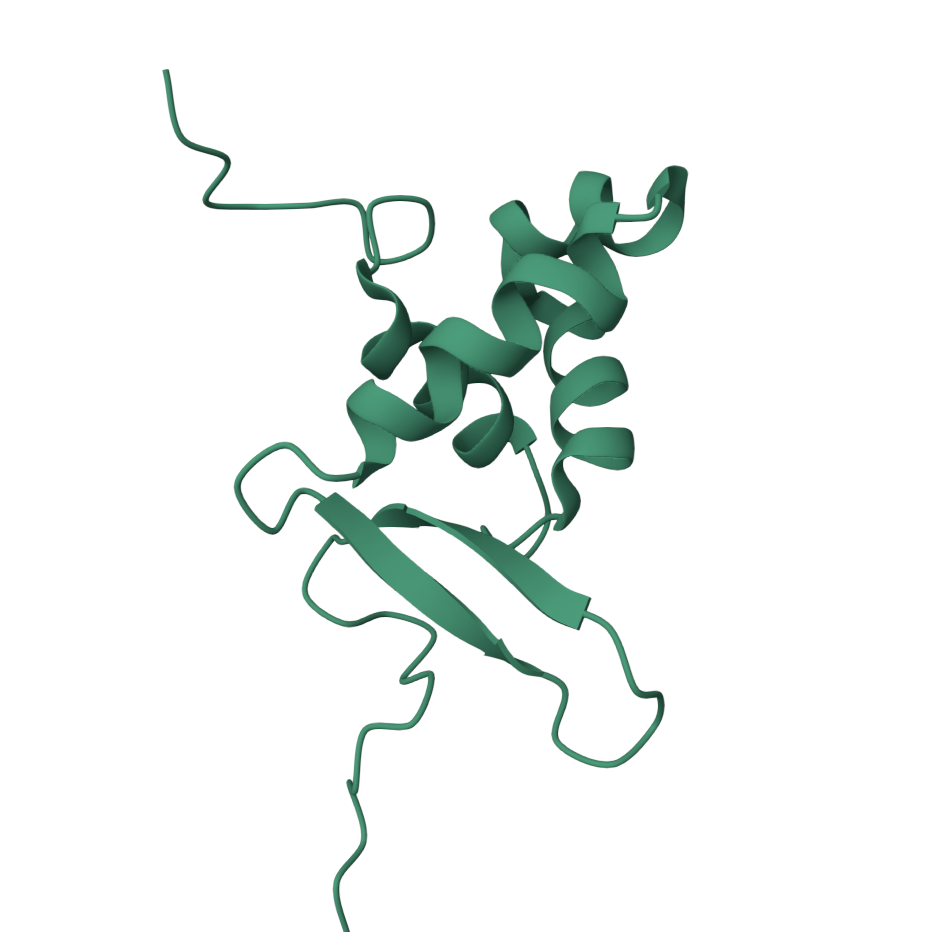
1 Briefly describe the protein you selected and why you selected it.
FOXO3 for its suspected longevity benefits. Certain alleles are found at higher frequencies in supercentenarians.
2 Identify the amino acid sequence of your protein.
• How long is it? What is the most frequent amino acid?
673 amino acids (states in the UniProt page)
• How many protein sequence homologs are there for your protein?
250 homologs
• Does your protein belong to any protein family?
”member of the forkhead box class O (FOXO) subfamily” (from RCSB)
3 Identify the structure page of your protein in RCSB
• When was the structure solved? Is it a good quality structure?
The entry was submitted in 2008.
(page: https://www.rcsb.org/structure/2K86)
(I couldn’t find this on the page, so I asked AI for help and it said: “Experimental method is solution NMR, so there is no crystallographic resolution value like “2.7 Å”; resolution only applies to diffraction/EM, not to NMR”)
• Are there any other molecules in the solved structure apart from protein?
There’s no other molecules present (according to this page).
• Does your protein belong to any structure classification family?
Under “family & domains” in UniProt it says “Winged helix-like DNA-binding domain superfamily/Winged helix DNA-binding domain 1 hit”
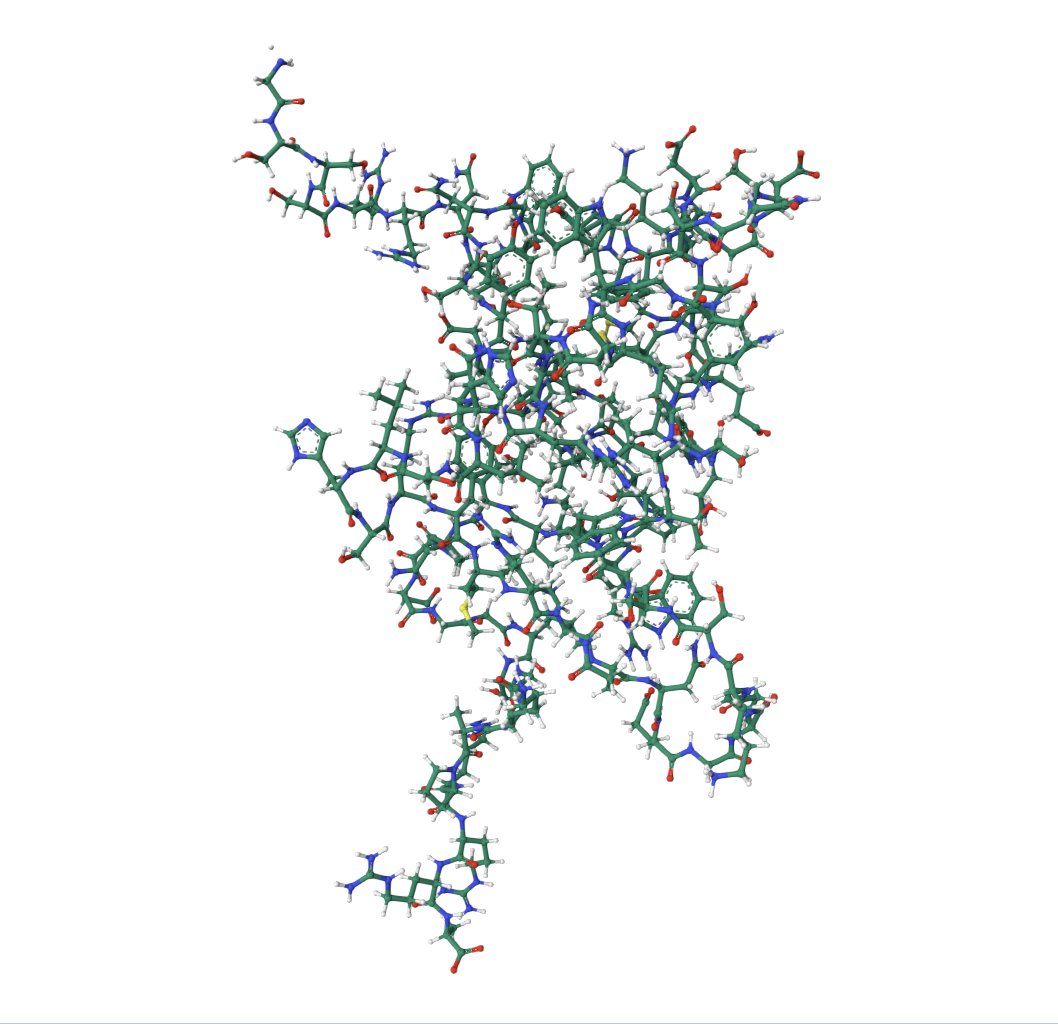
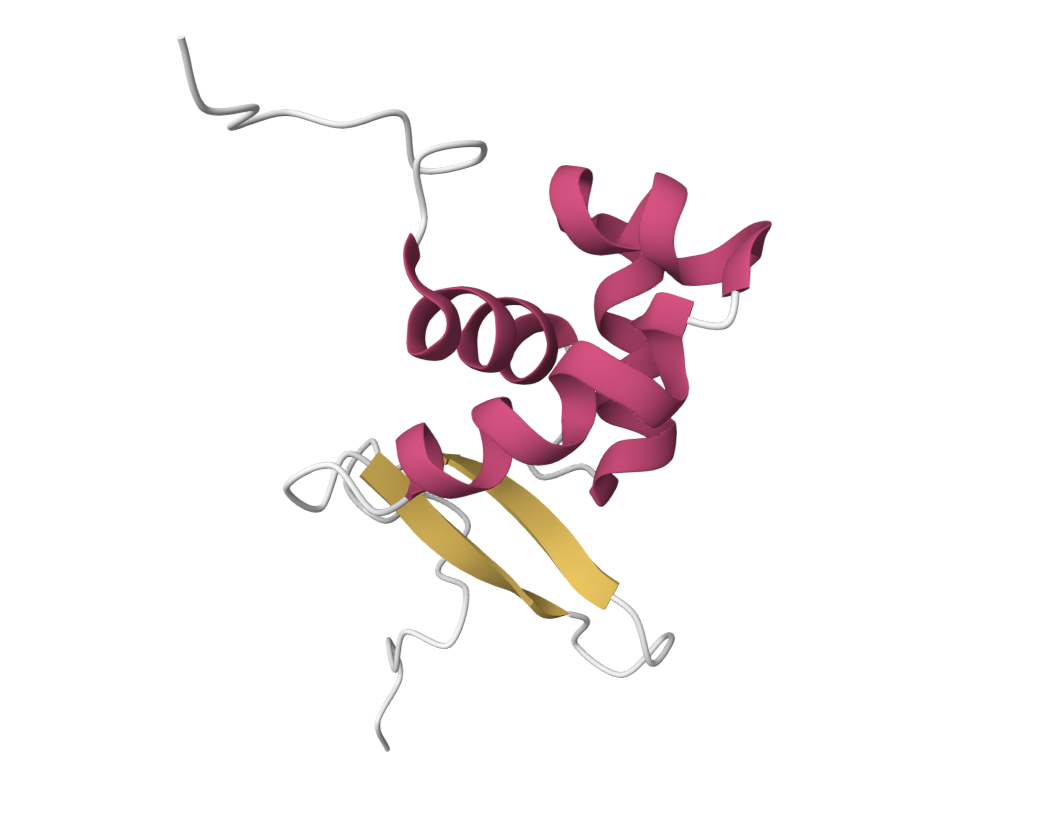
4 Open the structure of your protein in any 3D molecule visualization software:
• Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
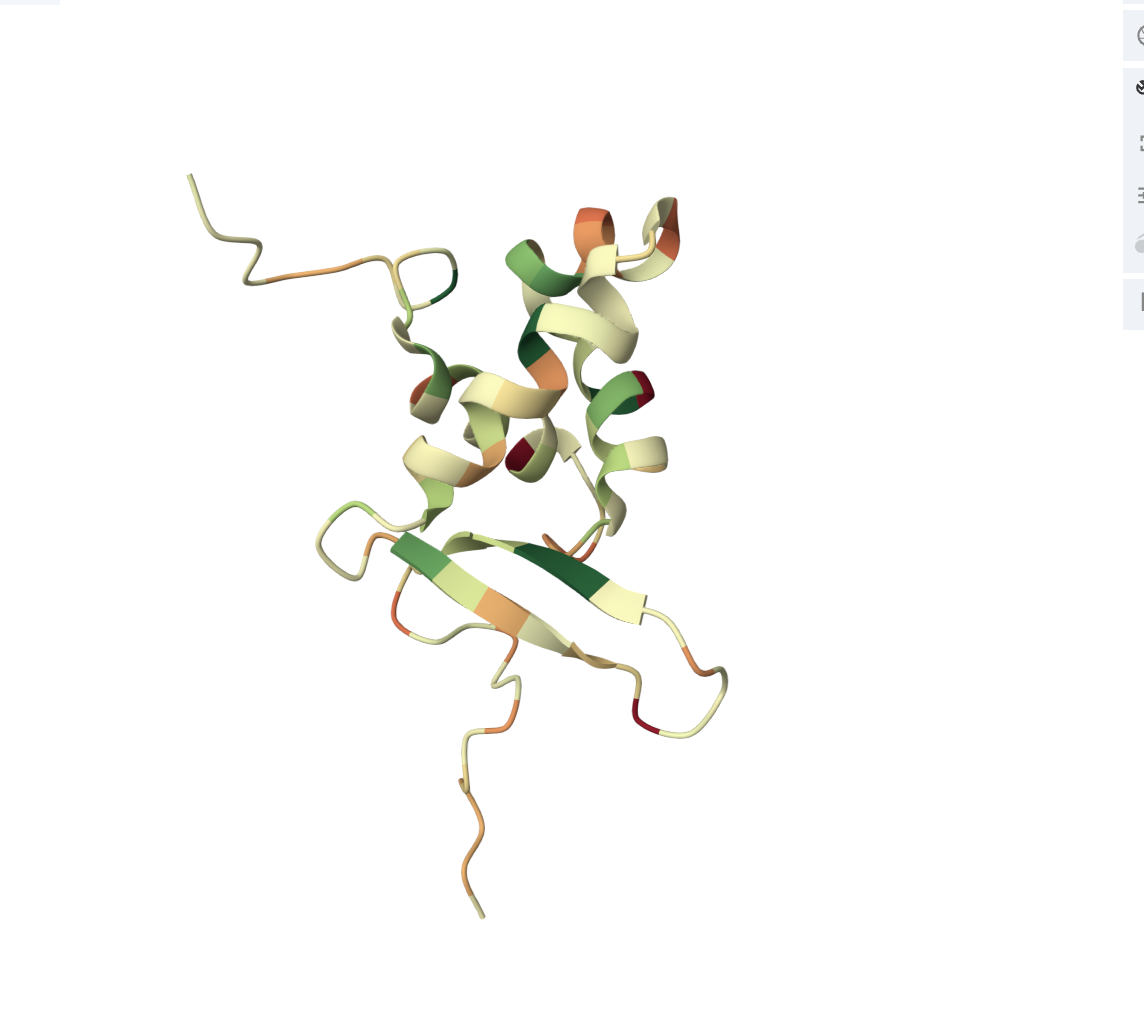
• Color the protein by secondary structure. Does it have more helices or sheets?
It has more helices.
• Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The core of the protein (interior) is dominated by hydrophobic residues (green patches). This is the hydrophobic core, which drives protein folding (hydrophobic residues hide from water by burying inward).
The outer loops and exposed regions show more orange/red, meaning hydrophilic/charged residues (these face outward toward the watery cellular environment).
• Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
💡 C1. Protein Language Modeling
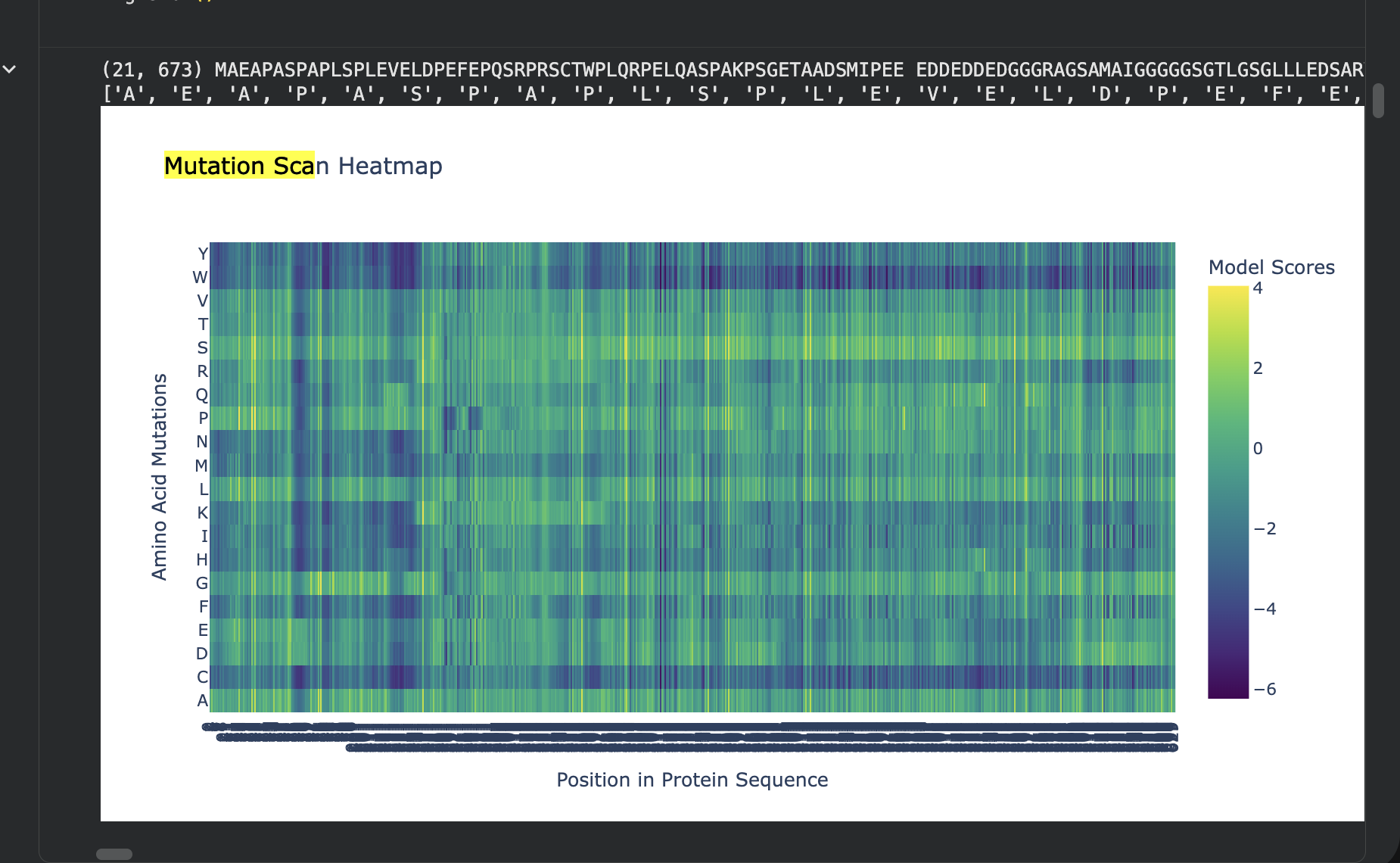
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The most disfavored mutation is:
Pos 1: M->W, LLR=-17.779
The most favored one:
Pos 261: T->S, LLR=3.991
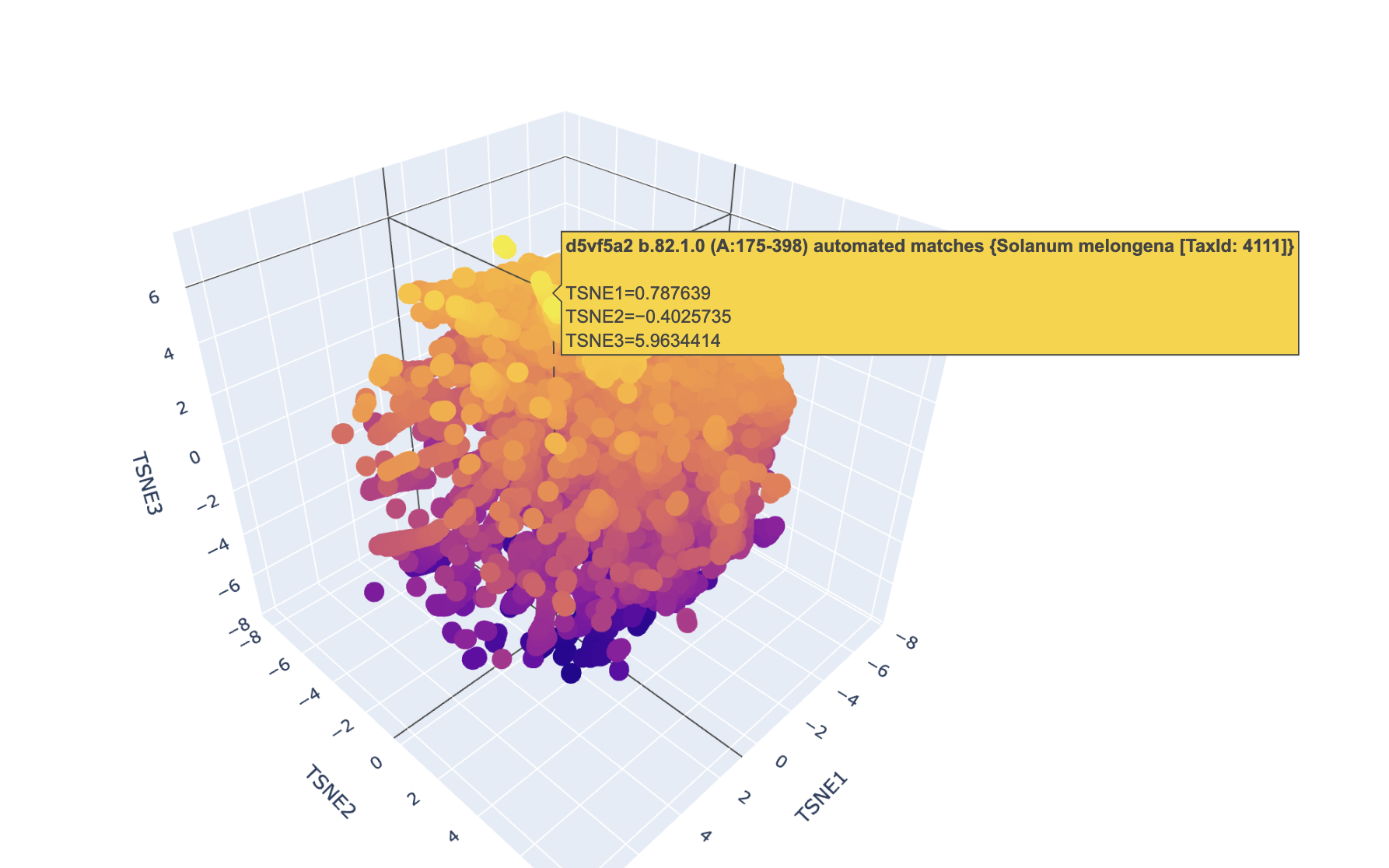
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
I had trouble finding this protein due to its nature.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
Week 5 HW: Protein Design Part II
🔎 Part 1: Generate Binders with PepMLM
Sequence with mutation:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
![[htgaa-hw5-pt1-binders.png]]
In text, the generated ones (plus the one provided to compare) were:
AlphaFold however was throwing errors with these sequences, so I asked Claude to check:
![[Screenshot 2026-03-08 at 17.05.41.png]]
So I changed them to these:
WRYYAAALRHKG
WRYYAVAARHKK
WRSYVVVLELGG
HHYPAVAVALKG
FLYRWLPSRRGG
🔎 PeptiVerse results
🦠 Peptide #1: WRYYAAALRHKG
AlphaFold:
ipTM = 0.38
pTM = 0.8
![[Screenshot 2026-03-08 at 18.17.58.png]]
![[Screenshot 2026-03-08 at 19.08.57.png]]
➡️ Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
ipTM = 0.38 is low confidence of binding. It would be surface-bound and far from the beta-barrel region.
PeptiVerse:
Input
⋮
Property
Prediction
Value
Unit
WRYYAAALRHKG
💧 Solubility
Soluble
1.000
Probability
WRYYAAALRHKG
🩸 Hemolysis
Non-hemolytic
0.016
Probability
WRYYAAALRHKG
🔗 Binding Affinity
Weak binding
6.017
pKd/pKi
WRYYAAALRHKG
📏 Length
12
aa
WRYYAAALRHKG
⚖️ Molecular Weight
1491.7
Da
WRYYAAALRHKG
⚡ Net Charge (pH 7)
2.84
WRYYAAALRHKG
🎯 Isoelectric Point
10.28
pH
WRYYAAALRHKG
💦 Hydrophobicity (GRAVY)
-0.90
GRAVY
🦠 Number #2 WRYYAVAARHKK
Alphafold:
ipTM = 0.31
pTM = 0.75
![[Screenshot 2026-03-08 at 18.19.06.png]]
➡️ Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Surface-bound, relatively close to the mutation site. It does not engage the beta barrel site. iPTM = 0.31 which shows low confidence.
PeptiVerse:
Input
⋮
Property
Prediction
Value
Unit
WRYYAVAARHKK
💧 Solubility
Soluble
1.000
Probability
WRYYAVAARHKK
🩸 Hemolysis
Non-hemolytic
0.017
Probability
WRYYAVAARHKK
🔗 Binding Affinity
Weak binding
5.699
pKd/pKi
WRYYAVAARHKK
📏 Length
12
aa
WRYYAVAARHKK
⚖️ Molecular Weight
1548.8
Da
WRYYAVAARHKK
⚡ Net Charge (pH 7)
3.84
WRYYAVAARHKK
🎯 Isoelectric Point
10.45
pH
WRYYAVAARHKK
💦 Hydrophobicity (GRAVY)
-1.16
GRAVY
🦠 Number #3 WRSYVVVLELGG
Alphafold:
ipTM = 0.75
pTM = 0.86
![[Screenshot 2026-03-08 at 18.48.53.png]]
➡️ Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
This one is bound on the surface. ipTM = 0.75 which is somewhat confident (we treat a score between 70 and 90 as “confident”). It is somewhat close to the beta-barrel area. It isn’t too far from the mutation area, but also not super close.
![[Screenshot 2026-03-08 at 18.54.30.png]]
PeptiVerse:
Input
Property
Prediction
Value
Unit
WRSYVVVLELGG
💧 Solubility
Soluble
0.999
Probability
WRSYVVVLELGG
🩸 Hemolysis
Non-hemolytic
0.113
Probability
WRSYVVVLELGG
🔗 Binding Affinity
Weak binding
6.406
pKd/pKi
WRSYVVVLELGG
📏 Length
12
aa
WRSYVVVLELGG
⚖️ Molecular Weight
1377.6
Da
WRSYVVVLELGG
⚡ Net Charge (pH 7)
-0.24
WRSYVVVLELGG
🎯 Isoelectric Point
6.00
pH
WRSYVVVLELGG
💦 Hydrophobicity (GRAVY)
0.70
GRAVY
🦠 Number 4: HHYPAVAVALKG
Alphafold:
ipTM = 0.29
pTM = 0.86
![[Screenshot 2026-03-08 at 18.15.35.png]]
➡️ Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
ipTM = 0.29 which is very low confidence of binding. No, it’s not close to the mutation site. It’s on the opposite side of there the beta-barrel sits, so no. It is surface-bound and closest to the positions 86-87.
PeptiVerse:
Input
⋮
Property
Prediction
Value
Unit
HHYPAVAVALKG
💧 Solubility
Soluble
1.000
Probability
HHYPAVAVALKG
🩸 Hemolysis
Non-hemolytic
0.048
Probability
HHYPAVAVALKG
🔗 Binding Affinity
Weak binding
5.251
pKd/pKi
HHYPAVAVALKG
📏 Length
12
aa
HHYPAVAVALKG
⚖️ Molecular Weight
1262.5
Da
HHYPAVAVALKG
⚡ Net Charge (pH 7)
0.93
HHYPAVAVALKG
🎯 Isoelectric Point
8.61
pH
HHYPAVAVALKG
💦 Hydrophobicity (GRAVY)
0.33
GRAVY
🦠 Number 5:FLYRWLPSRRGG
Alphafold:
ipTM = 0.35
pTM = 0.8
![[Screenshot 2026-03-08 at 18.20.14.png]]
➡️ Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
ipTM = 0.35, hence confidence of binding is low. It’s far from the N-terminus that has the A4V. It is surface-bound. It is close to the beta-barrel region (the arrows).
PeptiVerse:
Input
Property
Prediction
Value
Unit
FLYRWLPSRRGG
💧 Solubility
Soluble
1.000
Probability
FLYRWLPSRRGG
🩸 Hemolysis
Non-hemolytic
0.047
Probability
FLYRWLPSRRGG
🔗 Binding Affinity
Weak binding
5.968
pKd/pKi
FLYRWLPSRRGG
📏 Length
12
aa
FLYRWLPSRRGG
⚖️ Molecular Weight
1507.7
Da
FLYRWLPSRRGG
⚡ Net Charge (pH 7)
2.76
FLYRWLPSRRGG
🎯 Isoelectric Point
11.71
pH
FLYRWLPSRRGG
💦 Hydrophobicity (GRAVY)
-0.71
GRAVY
🔎 Part 1 analysis
➡️ In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Yes, WRSYVVVLELGG has a higher iPTM (0.75 vs 0.35) plus a lower perplexity score (18 vs 20), even though it has the highest perplexity score out of the other generated ones.
In general, the iPTM values for all of them except this one are low (~0.30), meaning that AlphaFold is not very confident about their vinding affinity.
➡️ In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
![[Screenshot 2026-03-08 at 19.22.45.png]]
WRSYVVVLELGG scores the highest iPTM from AlphaFold and the highest binding affinity according to PeptiVerse. The scores are correlated in both platforms for all peptides.
Solubility is around 1.0 for all of them, so none are poorly soluble.
All show hemolytic probabilities under 0.1 besides one, which is the strongest binder. The strongest binder is also one of the only two with a positive GRAVY score, showing the strongest hydrophobicity.
(I learned that “Hydrophobic peptides often damage membranes”, so this would not be a good candidate). Therefore the binder with the strongest affinity also shows the highest toxicity and aggregation risk.
Peptide #1 WRYYAAALRHKG shows the lowest hemolysis score, but its binding affinity is not too strong, so it would not make for a strong candidate, but it has a good safety profile.
Peptide #2 has weak binding affinity and a high charge, so we discard it.
(Why: “High overall positive charge is usually bad for a peptide drug candidate because it increases nonspecific binding to negatively charged membranes and biomolecules, which can drive toxicity, hemolysis, and off‑target effects”)
➡️ Choose one peptide you would advance and justify your decision briefly. I would choose peptide #1 because it has the second highest binding affinity (despite still being moderately low) but a good safety profile.
Week 6 HW: Genetic Circuits Part I
SECTION 1: PCR & GIBSON ASSEMBLY
“What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
It is composed of 3 essential components:
1. Phusion DNA Polymerase: a high-fidelity enzyme with low error rates (50x higher fidelity than Taq) and fast extension speeds (15-30sec/kilobase)
2. dNTPs (deoxynucleotide triphosates) (dATP, dCTP,dGTP, dTTP): the nucleotide building blocks the polymerase incorporates into the DNA starnd it builds
3. An optimized reaction buffer containing Mg(^2+) ions (polymerase cofactor that also stabilizes annealing) and KCl, which promotes specific primer binding while suppressing non-specific interactions.
The Green Master Mix also contains other reagents (green dye, density reagents) so the reaction can be directly loaded into an agarose gel.
What are some factors that determine primer annealing temperature during PCR?
It is determined by the melting temperature (Tm) of the primer, which is influenced by factors such as the primer length (longer = higher Tm), the GC content (more GC = higher Tm), and salt concentration in the reaction buffer.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
They are both methods for generating linear DNA fragments, with different strengths.
PCR is used to exponentially amplify a target region through cyclic multi-step iterations (denaturation -> annealing -> extension). It uses sequence-specific primers + a thermostable DNA polymerase to produce DNA fragments that match the primer binding sites, and can introduce mutations or add overlapping sequences (like the ones used in Gibson assembly). It is especially useful when we want to generate much larger quantities of a small sample of DNA.
Restriction enzyme digestion is preferred where fidelity is critical, i.e. when generating sticky ends for traditional ligation cloning or working on large constructs.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
For this to be the case, the following conditions must be met:
Homologous overlaps must exist between adjacent fragments (the 3’ end of one fragment must share 20-40 bp of sequence identity with the 5’ end of the next fragment)
Template DNA must be removed
The fragments must be purified (to remove enzymes, primers and nucleotides that could interfere with the Gibson reactions)
The purified DNA concentration should be high enough to provide enough material for the Gibson reaction
How does the plasmid DNA enter the E. coli cells during transformation?
For example, in the case of the heat shock method (used in the lab for this module), we pre-treat the cells with CaCl_2 (to neutralize the negative charges on the phospholipid membrane and the DNA backbone) to create “(chemically) competent cells”.
We incubate both the cells and the plasmid DNA on ice for 30 minutes, then induce a 42’C heat shock for exactly 45 seconds to induce a thermal gradient which is believed to open transient pores in the cells’ membrane which allows the DNA to pass through, We finish by returning them to ice for 5 minutes so the membranes are sealed again, with the DNA now inside.
Lastly, we incubate the cells in SOC-rich medium for 60 minutes (at 37ºC with shaking) to allow them to recover and keep dividing. Selection is applied by expressing the antibiotic resistance gene (only the cells that have taken up the DNA will survive in the new media with antibiotic).
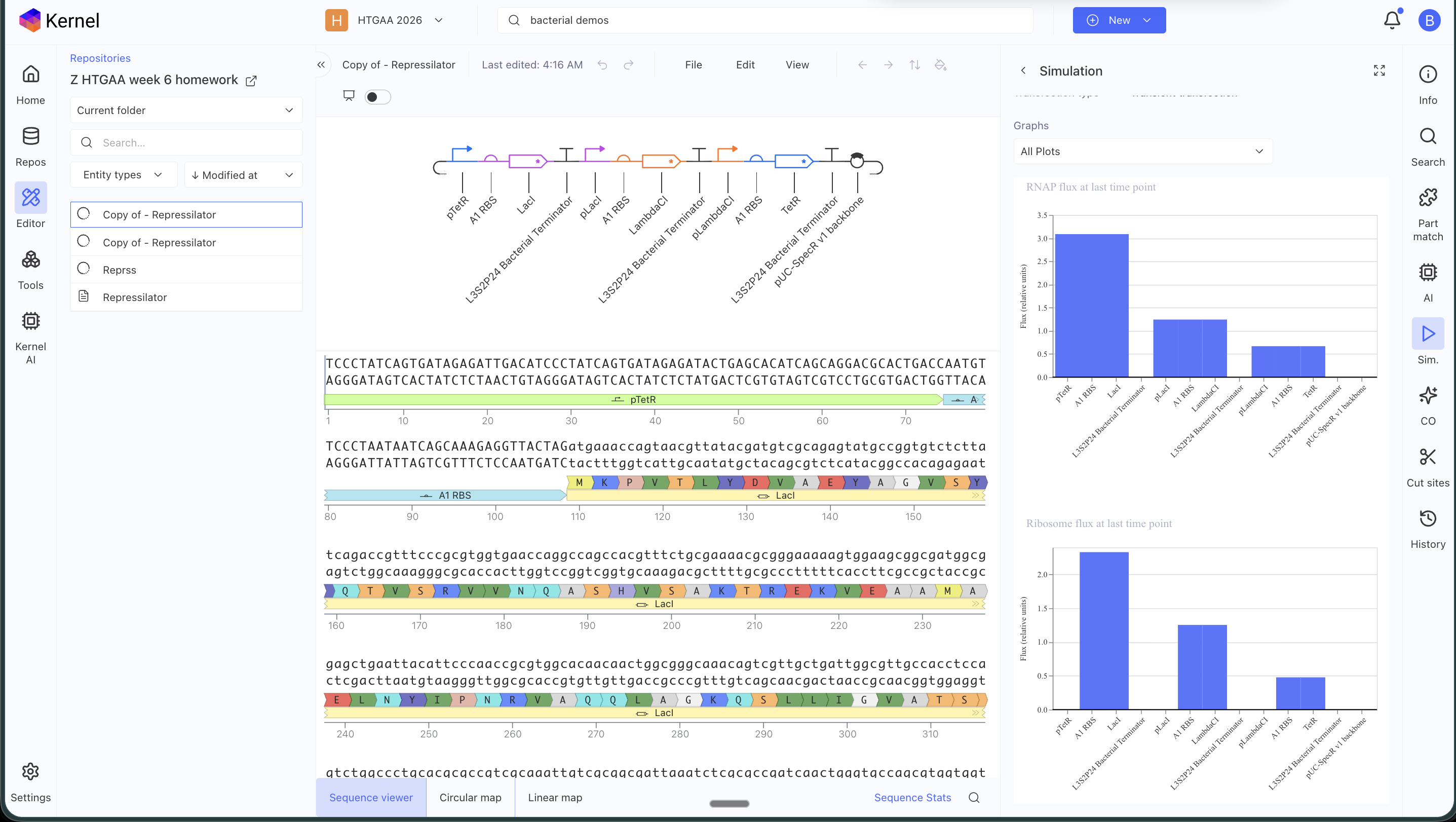
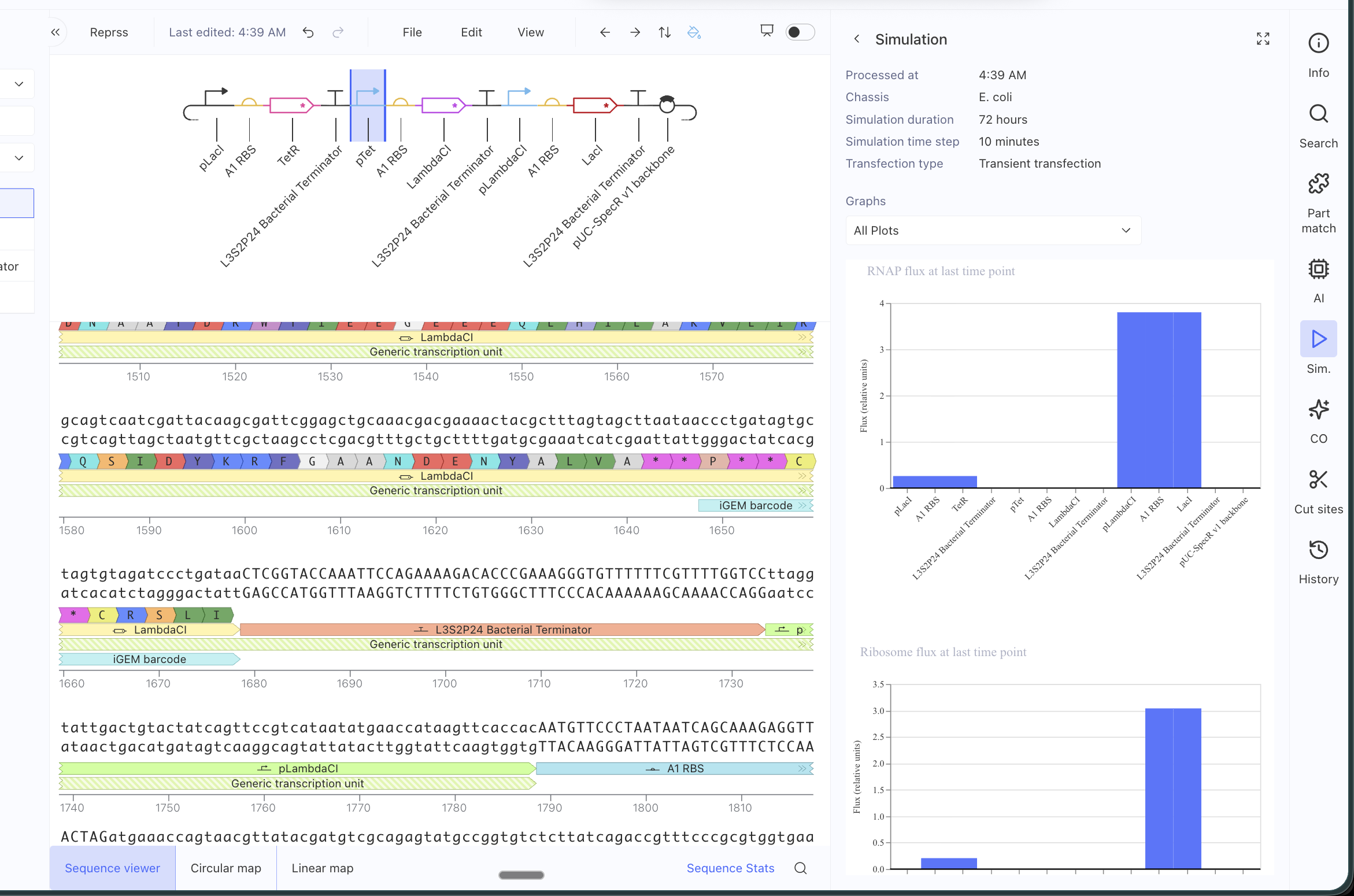
ASIMOV KERNEL REPRESSILATOR
Original repressilator construct from the example repo, with its simulation data:
My simulation graphs did not match:
Even after replacing all the parts specifically for the ones in the Characterized Bacteria repo, it still didn’t match. I will have to debug further.
I asked AI to help me debug and it said that some possible reasons might be that the parts I am using might have different metadata even if they appear identical, that my simulation window might be too long, that the RBS I chose might be too weak or strong, that the backbone or copy-number might differ (I tried with and without an added backbone), that the promoters might not have the right repressor behavior… many alternatives to test!
Week 7 HW: Genetic Circuits Part II
PART 0
1. How do ERNs work + how this differs from proteases
They cleave mRNA transcripts before they can be translated into proteins, so the mRNA is not processed by the ribosomes and gets degraded quickly by cellular exonucleases.
They work directly at the RNA level, unlike proteases which work on the final translated proteins. This means that they prevent new proteins from being produced, but they do not affect the existing pool of proteins.
ERNs recognize specific RNA sequences or structural motifs, while proteases recognize speciifc amino acid sequences (or structural features on folded proteins).
In summary: ERNs work at the pre-translational stage and detect specific RNA sequences or motifs sites that they cleave directly, while proteases act on the post-translational product guided by amino acid sequences or structural patterns.
2. Lipofectamine 3000 mechanism
Lipofectamine 3000 is a lipid-based transfection reagent that forms positively-charged lipid nanoparticles which bind to the negatively-charged plasmid DNA to create “lipoplexes”. These particles are then taken up by cells through endocytosis thanks to a similar mechanism, where the positively-charged lipoplex interacts with the negatively-charged cell membrane causing it to absorb it as an internal (endosomal) vesicle. They then “escape” the endosomal container by rupturing it (the lipids buffer the acidifying endosome), releasing the plasmid DNA into the cytoplasm before it can be degraded in lysosomes.
(Interestingly, I have been recently researching mRNA-LNP vaccines and they use a similar mechanism for “endosomal escape”!)
The DNA must then enter the nucleus, where it will be recognized by the host RNA polymerase II, which will transcribe it into mRNA (which will in turn be translated into the final protein once exported to the cytoplasm and processed by ribosomes).
3. Poly-transfection + why useful in neuromorphic circuits
Poly-transfection is the process of transfecting multiple plasmids into the same population of cells (so each cell receives and expressed multiple different genetic instructions). This is usually done by mixing all the target plasmids within the same lipofectamine reaction (the cell uptake of the plasmids can be uneven, so the final sample will be heterogeneous).
This is important for neuromorphic circuits because they require a variety of functional parts that could not fit into a single plasmid, due to size constraints and promoter interference (ie sensors, signal-processing nodes, output reporters). Some of these parts must be included within the same cell to operate as a singular functional/“computing” unit (like in a neural network).
PART 1 - IANNs
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional Boolean genetic circuits produce discrete and binary outputs (“on” or “off”), and therefore cannot represent continuous values or perform weighted computations.
IANNs can process continuous values as input and produce graded outputs, so they can be used to perform additional functions like weighted summation, thresholding and signal integration.
They are also more flexible over time, as their weights can be tuned and upgraded over time through poly-transfection, which means that the same circuit can be adapted for different use cases.
Lastly, they are more compact, as a single IANN can handle calculations that would require multiple Boolean circuits to compute.
(An analogy I liked is that traditional genetic circuits are like ON/OFF switches, while IANNs are more like dimmer switches with memory, which can represent a value between 0-100%)
Their ability to handle non-discrete values in computation makes them a much better fit for computations in biological environments (which are noisy in nature), since they can tolerate stochastic fluctuations (introducing noise won’t necessarily lead to a false negative or positive, since the final output is graded, instead of a “yes/no”).
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
We could use IANNs to build a cell-state classifier, which could distinguish between states (ie healthy, stressed or apoptotic) based on expression levels of different biomarker proteins (the inputs).
We would engineer the cells to have promoters that are sensitive to these specific proteins, and engineer a “hidden layer” that would compute weighted combinations that would act as signals for the output layer to express different fluorescent proteins to indicate each state (ie different colors for different states).
Example:
Possible cell states: healthy, stressed, apoptotic
Inputs: biomarker proteins (ie p53, caspase-3, which indicate apoptosis)
Computation: 1. promoter detects biomarkers 2. hidden layer computes weighted combinations 3. output layer expresses different proteins based on the received values
Output: signal proteins expressed (ie GFP for “healthy”, RFP for “stressed”, BFP fpr “apoptotic”)
The main difficulties would be the cell-to-cell variability after poly-transfection (explained in the relevant section above), which could introduce a lot of noise, and setting the correct weights (promoter strengths) to achieve classification accuracy.
Week 9 HW: Cell-Free Systems
Week 9 Homework: Cell-Free Systems
[WIP!]
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
The benefit can be summarized as “it removes the constraints we usually face for protein synthesis when working with live cells”. For example, working with live cells requires culturing cells throughout the whole cell lifecycle, with all the possibilities for error that this implies (mistakes, unexpected cell behavior due to their non-deterministic nature, etc), as well as the required timelines (speed is limited by the fundamental speed constraints from the cell growth cycle), and costs.
Cell-free systems also allow for much greater control, as the main system is boiled down to its most basic functional components, removing a lot of complexity (variables outside of our control), and therefore allowing the possibility of producing much more homogeneous products.
Two special cases that CFS allow are the use of molecules that would usually be toxic for cells, and working with a much wider variety of molecules that would otherwise be destroyed by the cell, as well as modified versions of the most basic components of this machinery (that could not work in a live cell), such as amino acids that don’t exist in nature.
Describe the main components of a cell-free expression system and explain the role of each component.
The basic components are:
1) The lysate, which includes the “machinery”: basically ground up cells (prokaryotic or eukaryotic), providing the cellular components we need for the reactions, like ribosomes, enzymes, tRNAs and cofactors, which will translate mRNA into protein
2) The genetic template (DNA or RNA) that encodes the product we want
3) The “building blocks” (amino acids and nucleotides)
4) The energy system (needed to provide and replenish the ATP and GTP needed for the reactions) (ie Mg^2+ and K+)
1) Note: CFS use Phosphoenolpyruvate (PEP) for this
5) The “environmental tuning” factors (salts, buffers, temperature, etc)
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Because protein synthesis is an energy-intensive process. In living cells, mitochondria handle the production of ATP, but in CFS we need to set up systems that can continually obtain ie the phosphates needed to convert ADP into ATP.
Not being able to meet these energy needs will cause the reactions to stall or provide low yield.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Only eukaryotic cells can perform PTM post-translational modifications (ie glycosylation), so any desired product requiring this process will require the use of eukaryotic cell lysate. This will also be required if the final desired product requires the usage of mammalian regulators.
However, when these requirements are absent, prokaryotic systems are faster and cheaper.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The main challenge when working with membrane proteins is that they tend to aggregate or misfold in aqueous environments (due to their hydrophobic regions trying to “escape” it) [ PENDING ]
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
The required energy needs are not being met.
The proteins might be misfolding. In this case, we would check and tune the environmental variables, like the temperature or the salsts, and possibly add chaperones or use different membrane-mimicking systems.
The final or intermediary products might be suffering degradation. In this case, I would check for the presence of ie nucleases, proteases, etc.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Could this function be realized by genetically modified natural cell?
Describe the desired outcome of your synthetic cell operation.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
What would you encapsulate inside? Enzymes, small molecules.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
How will you measure the function of your system?
PENDING
Example solution
Based on: Lentini, R. et al., 2014. Nat comm, 5, p.4012.
Pick a function and describe it.
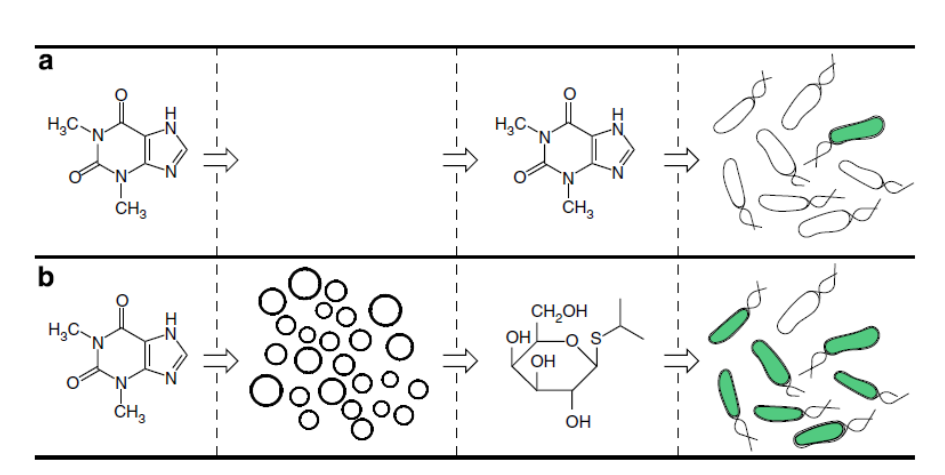
What would your synthetic cell do? What is the input and what is the output? Expand the sensing capacity of bacteria. Input: theophylline (inert to bacteria). Output of the SMC: IPTG. Output of the whole system: GFP produced in bacteria. (Theophyline aptamer reference: Martini, L. & Mansy, S.S., 2011. Cell-like systems with riboswitch controlled gene expression. Chemical Communications, 47(38), p.10734.)
Could this function be realized by cell-free Tx/Tl alone, without encapsulation? No. If the IPTG were not encapsulated, it would go into the bacteria without the need of theophylline-induced membrane channel synthesis, thus the synthetic cell actuator would not exist.
Could this function be realized by genetically modified natural cell? Yes, in this particular case: the theophylline aptamer could be incorporated into a transformed gene. This lacks generality though – it is easier to make SMC than modify bacteria, so in this system a single bacteria reporter can be used to detect various small molecules.
Describe the desired outcome of your synthetic cell operation. In the presence of SMC, bacteria sense theophylline.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of? Phospholipids + cholesterol.
What would you encapsulate inside? Enzymes, small molecules. cell-free Tx/Tl system, IPTG, gene for membrane transporter under the control of theophylline aptamer.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian) Bacterial, because of the theophylline riboswitch used as SMC input.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?) The membrane is permeable to the input molecule (theophylline), the output is IPTG that will cross the membrane via the membrane pore created after theophyline-initiated gene expression.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: POPC, cholesterol
Enzymes: bacterial cell-free Tx/Tl
Genes: a-hemolysin (aHL) to encapsulate in SMC
Biological cells: E.coli transformed with GFP under T7 promoter and a lac operator
How will you measure the function of your system? Measure GFP output of the cells via flow cytometry. Alternatively, use enzymatic reporter, like luciferase, and measure bulk output of the enzyme.
Artificial cells translate chemical signals for E. coli. (a) In the absence of artificial cells (circles), E. coli (oblong) cannot sense theophylline. (b) Artificial cells can be engineered to detect theophylline and in response release IPTG, a chemical signal that induces a response in E. coli.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
How will the idea work, in more detail? Write 3-4 sentences or more.
What societal challenge or market need will this address?
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
MY ANSWERS:
Portable FD-CF pathogen sensor patches embedded in taxi and public transport vehicles to monitor airborne pathogen load
Leveraging a freeze-dried system with an RNA switch (or CRISPR-based) detector, this device could be carried by transport service providers who could periodically measure the pathogen levels in the vehicle with a swab. It could be integrated into seatbelts or any other part of the car (it could be a patch or sticker).
Use cases: 1) identify when the vehicle should be more thoroughly disinfected 2) driver safety 3) passenger safety (for example, riders who are especially immunocompromised could request vehicles with lower pathogen levels) 4) data collection for scientific purposes (ie epidemic detection and prevention)
The device could be a patch with a cartridge containing the necessary water and buffers and a button or switch that the driver could click to easily mix the components, for example at the end of a day’s shift. The device would be single-use and replaced daily to ensure minimal contamination.
The freeze-dried system ensures stability for at least 3-6 months at room temperature.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
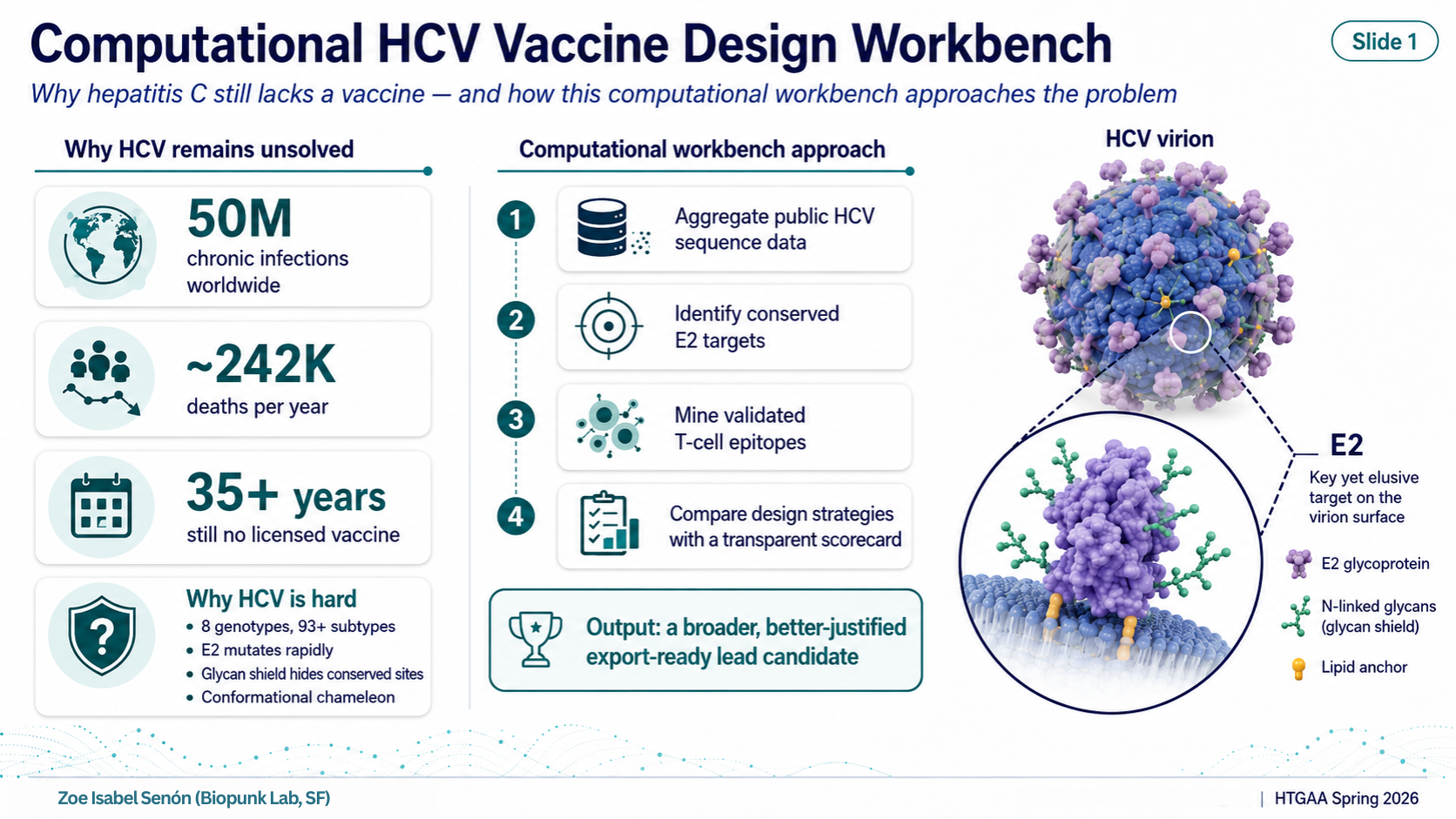
Computational HCV Vaccine Design Workbench HTGAA Spring 2026 — Final Project What Is This Project? In one line: a reproducible workbench that nominates an annotated HCV immunogen construct for future experimental validation.
In one line: a reproducible workbench that nominates an annotated HCV immunogen construct for future experimental validation.
A fully computational pipeline that collects public hepatitis C virus (HCV) sequence data, identifies the most conserved and immunologically validated regions of the viral envelope, designs a family of candidate immunogens using different strategies, scores each candidate against a transparent multi-criterion rubric, and exports the top-ranked construct as an annotated, codon-optimized DNA and protein sequence ready for experimental testing.
The Problem It Addresses
Hepatitis C virus (HCV) has infected roughly 50 million people worldwide and kills around 242,000 per year (WHO 2024). Despite more than 35 years of research, there is no licensed vaccine.
The core obstacle is the E2 envelope protein — the part of the virus that binds host cells and is the primary antibody target. E2 is a difficult target for three reasons:
Extreme genetic diversity. HCV has 8 confirmed genotypes and 93+ subtypes (ICTV 2026). A vaccine designed against a single reference strain trains the immune system on one variant of a constantly shifting target.
The glycan shield. E2 carries 9–11 N-linked glycosylation sites that act as a molecular camouflage, physically blocking antibody access to the conserved surfaces that would actually be worth targeting.
Conformational instability. E2 adopts different shapes depending on context. Linear sequence-level approaches cannot fully capture the neutralizing epitopes that only exist as three-dimensional structures.
Computational approaches can help by aggregating diversity data, identifying what is conserved despite the variability, and systematically comparing design strategies before committing to synthesis or animal studies.
The Pipeline — 7 Stages
Stage 1 — Sequence Collection
Public HCV E2 protein sequences downloaded from NCBI Virus. A curated panel spanning four genotypes (1a, 1b, 2a, 3a) was assembled, covering an estimated 65–75% of global HCV infections. All sequences are logged in sequence-manifest.tsv with accession numbers and source metadata.
Stage 2 — Multiple Sequence Alignment
All sequences aligned using MAFFT v7, producing a column-level alignment. Alignment quality was verified manually. Output: aligned FASTA, 434 columns.
Stage 3 — Conservation Analysis
Per-position Shannon entropy calculated in Python across all alignment columns. A 15-amino-acid sliding window was used to identify contiguous conserved regions. The most conserved window (positions 297–311, overlapping the CD81 binding loop) reached 99.15% conservation. Mean conservation across all positions: 87.2%.
Stage 4 — Epitope Mining
The Immune Epitope Database (IEDB) was queried for positive T-cell assay results from HCV NS3 and NS5B proteins in human hosts. Results were ranked by a composite score weighting assay count, number of independent publications (PMIDs), HLA type count, and population coverage. Top hits: KLVALGINAV (NS3, 90 assays, 18 PMIDs, HLA-A*02:01), CINGVCWTV (NS3, 57 assays, 17 PMIDs), ALYDVVTKL (NS5B, 4 assays), IMAKNEVFCV (NS5B, 4 assays).
Stage 5 — Construct Design
Five candidate immunogens were designed:
Design
Strategy
Length
A — Natural Baseline
Single genotype 1a reference (strain H77)
420 aa
B — Consensus E2
Statistical consensus of all curated sequences
434 aa
C — Hybrid E2 + T-cell
Consensus E2 fused to 4 validated NS3/NS5B T-cell epitopes
492 aa
D — E2-Ferritin Nanoparticle
Consensus E2 displayed on H. pylori ferritin 24-mer scaffold
640 aa monomer / ~1.69 MDa particle
D-2 — Focused Conserved Mosaic
Only the top conserved E2 windows + T-cell cassette
97 aa
Stage 6 — Multi-Criterion Scoring
Each design scored on a 100-point, 6-axis rubric:
Criterion
Weight
What It Measures
Genotype Coverage
25
Breadth of genotype representation
Epitope Conservation
20
Per-position amino acid identity at functional sites
HLA Coverage
15
Population coverage of T-cell epitopes
Developability
15
Expression feasibility, folding confidence
Manufacturability
15
Construct length, GC content, synthesis cost
Interpretability
10
Clarity and communicability of design rationale
Final scores: A = 45, B = 66, C = 76, D = 68, D-2 = 89.
Stage 7 — Export
Top candidates exported as: codon-optimized DNA FASTA, protein FASTA, construct map, annotations TSV. Design C codon optimization: GC content reduced from 68.6% → 47.6% (into the mRNA synthesis sweet spot of 40–60%).
Tools Used
Tool
Purpose
NCBI Virus
Public HCV E2 sequence retrieval
MAFFT v7
Multiple sequence alignment
Python (Biopython, NumPy)
Shannon entropy, sliding window conservation, FASTA handling
IEDB (Immune Epitope Database)
T-cell epitope mining and ranking
ESMFold
Protein structure confidence prediction (pLDDT scoring)
Python codon optimization script
Human codon-usage optimization, GC content balancing
Custom scoring framework
6-axis, 100-point scorecard (documented in design-scorecard.draft.tsv)
All tools are public and free. The pipeline is fully scripted in Python and re-runnable.
The Lead Nomination — Design D: E2-Ferritin Nanoparticle
Architecture: Consensus E2 (the full 434-residue sequence representing the statistical average across all curated sequences) genetically fused via a flexible (G4S)₃ linker to H. pylori ferritin (UniProt P52093, 167 aa). The monomer is 640 aa and self-assembles into a 24-subunit octahedral nanoparticle of approximately 1.69 MDa.
Why ferritin display matters: Presenting 24 copies of the same antigen on a symmetric scaffold drives B-cell receptor cross-linking — the same signal a real virus surface sends. This mechanism has empirical precedent: Kanekiyo et al. 2013 (Nature 499:102–106) demonstrated approximately a 10-fold increase in neutralizing antibody titers when the influenza HA-stem antigen was displayed on the same H. pylori ferritin scaffold compared to soluble antigen. The scaffold itself is well-characterized and the particle size (~1.69 MDa, ~10–15 nm) is in the range optimal for lymph node trafficking and B-cell engagement.
The scorecard paradox: D-2 scores 89/100 because it is small (97 aa), easy to synthesize, and carries a full T-cell epitope cassette. Design D scores lower because size (640 aa) reduces manufacturability points and the scorecard has no axis for multivalent avidity — that biological multiplier is invisible to sequence-level scoring. The scorecard is a decision-support tool, not an efficacy oracle. It captures what can be computed from sequence. The Kanekiyo precedent captures what biology does with geometry.
The Scorecard Runner-Up — Design D-2: Focused Conserved Mosaic
Design D-2 wins the raw scorecard at 89/100. It is a 97-amino-acid linear construct made entirely of: the two most conserved E2 windows (99.15% and 97.73% conservation respectively) connected by GGGGS linkers, plus the same four-epitope T-cell cassette.
It scores so high because it maximizes conservation efficiency (only top-1% positions included), retains full HLA coverage from the T-cell cassette, and is dramatically cheaper to synthesize than any of the full-length designs.
It does not lead because linear peptides stripped of their structural context rarely elicit neutralizing antibodies. The surfaces that broadly neutralizing antibodies recognize on E2 are conformational — they require the native fold. D-2 is the right candidate for T-cell peptide assays, for cost-limited settings, or for display ON the ferritin scaffold (a combined D+D-2 construct is the highest-priority future design).
Validation — What Was Done Computationally
Sequence-Level Validation
All accession numbers logged and traceable
Conservation calculated by Shannon entropy (standard method)
Top conserved windows cross-referenced against known structural data: positions 297–311 overlap the CD81 binding loop, the target of broadly neutralizing antibodies HEPC3, AR3C, and AR5A (Kong et al. 2013 Science; Flyak et al. 2018 Cell)
Epitope Validation
All T-cell epitopes sourced from IEDB positive human assays only
Top epitope KLVALGINAV: 90 independent assays, 18 peer-reviewed publications
HLA coverage estimated at A02 supertype (~44% US phenotypic coverage per Sidney et al. 2008)
Structure Prediction
Ferritin scaffold (Design D): ESMFold mean pLDDT 0.937, 91% of residues above 0.9 — high-confidence autonomous folding
Full E2 consensus: ESMFold pLDDT ~0.3 — low confidence, expected for a disulfide-rich glycoprotein (8+ disulfide bonds and N-linked glycans cannot be modeled by ESMFold). Crystal structures of related strains exist (PDB: 4MWF, 4WEB, 6MEJ) but our consensus sequence has not been structurally characterized
Codon Optimization
Design C DNA sequence codon-optimized for human expression using human codon frequency tables
GC content reduced from 68.6% to 47.6%, within the 40–60% mRNA synthesis sweet spot
Verified for absence of common restriction sites
Limitations — What Was Not Done
No experimental expression, purification, or binding assay
No immunization data, no efficacy claim
E2 structural context not validated for the consensus sequence — conformational epitopes may differ from reference structures
Genotype 2a is represented by a single sequence; genotypes 4–7 are absent (genotype 4 alone accounts for ~8–17% of global infections, dominant in Egypt and North Africa)
HLA coverage limited to the A02 supertype; other HLA supertypes not analyzed
CINGVCWTV Class II MHC presentation not confirmed (9-mer is Class I-typical; IEDB reports 4 HLA types but Class II binding is unverified)
Future Validation Plan
Phase 1 — Expression and Binding (3–6 months)
Gene-synthesize Design C codon-optimized DNA; express in HEK293 or CHO cells
Confirm protein by Western blot and size-exclusion chromatography
Test binding against broadly neutralizing antibody panel: HEPC3 (Flyak et al. 2018), AR3C (Kong et al. 2013), AR5A (Giang et al. 2012), AP33 (Owsianka et al. 2005)
T-cell ELISpot with HLA-A*02:01 donor PBMCs using D-2 peptides
Phase 2 — Computational Refinement (parallel)
ColabFold / AlphaFold2 with disulfide constraints for consensus E2 structural prediction
NetMHCpan 4.1 and NetMHCIIpan 4.1 for HLA coverage beyond the A02 supertype
Expand genotype panel to include genotypes 4–7
Design the combined candidate: conserved E2 windows (D-2) displayed on the ferritin nanoparticle (D) with T-cell cassette in the SpyTag/(G4S)₃ linker
Phase 3 — In Vivo Testing (aspirational)
Head-to-head mouse immunization: Designs C, D, and the combined candidate